Association between inflammation and cigarette smoking in cardiac remodeling after acute myocardial infarction
ORIGINAL RESEARCH ARTICLE
Association between inflammation and cigarette smoking in cardiac remodeling after acute myocardial infarction
Article Summary
- DOI: 10.24969/hvt.2022.361
- Page(s): 23-33
- CARDIOVASCULAR DISEASES
- Published: 14/12/2022
- Received: 11/09/2022
- Revised: 16/11/2022
- Accepted: 17/11/2022
- Views: 8394
- Downloads: 5441
- Keywords: cardiac remodeling, inflammation, interferons, myocardial infarction, smoking
Address for Correspondence: Cengiz Sabanoglu Department of Cardiology, Kirikkale High Specialization Hospital, Baglarbasi District, Ahmet Ay Street, 71300, Kirikkale, Turkey Phone: +90532 280 4856, Fax: +90318 234 0606,
E-mail: drchingiz23@gmail.com
Cengiz Sabanoglu1, Nilnur Eyerci2, Orhan Karayigit3, Mehmet Ali Felekoglu4, Emrullah Kiziltunc5, Omer Faruk Ates6
1Department of Cardiology, Kirikkale High Specialization Hospital, Kirikkale, Turkey
2Department of Medical Biology, Kafkas University Faculty of Medicine, Kars, Turkey
3Department of Cardiology, Ministry of Health Yozgat City Hospital, Yozgat, Turkey
4Department of Cardiology, Atakent Hospital, Yalova, Turkey
5Department of Cardiology, Gazi University Faculty of Medicine, Ankara, Turkey
6Department of Radiology, Sakarya University Faculty of Medicine, Sakarya, Turkey
Abstract
Objective: In this study, we aimed to investigate the relationship between smoking and adverse cardiac remodeling after ST-elevation MI (STEMI), and the association between smoking and inflammatory markers, including cytokine levels.
Methods: Forty-three patients admitted to the emergency department between June 2015 and June 2020 who were diagnosed with STEMI for the first time and underwent successful primary percutaneous coronary intervention were included in the study. Inflammatory markers (interferon (IFN)-α, -γ, -β, interleukin (IL)-6R-α, and soluble tumor necrosis factor receptor (sTNFR)-1,-2) were measured on the first day and two weeks post-MI. Left ventricular volume and functions were evaluated using cardiac magnetic resonance imaging at two weeks and six months post-STEMI. Adverse remodeling (AR) was defined as an increase in left ventricular (LV) end-diastolic volume >12%.
Results: The AR ratio (65% vs. 30.4%; p= 0.024) and the levels of each inflammatory marker on the first-day post-STEMI were higher in the smokers' group than in the non-smokers' group. Smoking (OR= 4.46; p= 0.032) and IFN-β (OR= 1.07; p=0.023) levels on the first-day post-MI were independent predictors of AR. Also, smoking (β(SE)= 8.96(2.74); p=0.002), increased neutrophil levels (β±SE= 1.72(0.66); p= 0.013) and increased LDL levels (β(SE) = 0.07(0.03); p=0.031) were independent predictors of elevated IFN-β levels.
Conclusion: Baseline inflammatory marker levels and incidence of AR post-STEMI were higher in smokers. Smoking can contribute to the development of AR by increasing the severity of inflammation at the onset of acute STEMI.
Key words: cardiac remodeling, inflammation, interferons, myocardial infarction, smoking
Introduction
Smoking is the primary preventable danger factor for cardiovascular and cerebrovascular disorders, increasing the risk of developing coronary heart disease by approximately two fold (1, 2). World Health Organization data have indicated that approximately 5 million deaths are associated with smoking annually (3). Smoking-related cardiac damage occurs via a few different mechanisms, including inflammation, hypertrophy, oxidative stress, angiogenesis, energy metabolism abnormalities, apoptosis, and gap junction remodeling (4), leading to a disturbance in heart size, mass, and geometry, resulting in cardiac dysfunction (5). Inflammation has a primary role in these processes.
An inflammatory response develops in tissues due to ischemia during the post-myocardial infarction (MI) period (6). Cardiac repair is related to the intensity of inflammatory response. In the case of an excessive inflammatory response, levels of reactive oxygen species increase abruptly via nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidase activity, causing severe oxidative tissue damage and adverse cardiac remodeling (AR) (7, 8).
Mild inflammatory or a strong anti-inflammatory post-MI response leads to mild oxidative damage without progressive impairment of cardiac structure and function (9). Previous studies have reported that smoking increases some pro-inflammatory cytokines (10). There are no comprehensive studies regarding the effects of smoking on the post-MI-enhanced inflammatory response.
However, the well-known effects of smoking, regardless of MI, such as increased blood pressure and heart rate, endothelial dysfunction, inflammation, and oxidative stress, are suggestive of enhanced post-MI inflammatory response in smokers at risk of developing AR (11-13).
Therefore, this study aimed to investigate the relationship between smoking and adverse cardiac remodeling after ST-elevation MI (STEMI), and the association between smoking and inflammatory markers, including cytokine levels.
Methods
Study population
This multi-center prospective study was conducted between June 2015 and June 2016. It was conducted in accordance with the Declaration of Helsinki (revised 2013), Good Clinical Practice Guidelines and approved by the local ethics committee (Decision no: 2013/106). Written informed permission was acquired from all patients. Assuming an alpha of 0.05, a power of 0.80, and a 30% estimated AR ratio in line with the former reports (14), the calculated sample size was at least 40 cases.
Considering the third universal definition of MI (15), 567 patients (>18 years of age) who were admitted to the emergency department due to first-time STEMI were evaluated. All patients underwent primary percutaneous coronary intervention (pPCI) within 12 hours of symptom onset and pPCI was organized according to the latest European Society of Cardiology (ESC) guidelines (16). Previous evidence suggests that daily rhythm can cause expression differences on inflammatory markers (17, 18). Therefore, 43 patients who admitted to the emergency department in the early hours of work (08:00-12:00 A.M) and whose blood samples at similar times were taken and patients meeting inclusion criteria were included in the analysis.
Patients with any mechanical complications (ventricular septal rupture, ventricular free wall rupture or cardiac tamponade, and papillary muscle rupture), those in cardiogenic shock (defined as systolic blood pressure ≤ 90 mmHg) or patients in need of hemodynamic support, patients who had previously had silent ischemia / infarction, patients with right coronary artery occlusion, those with any systemic inflammatory disease, those with autoimmune disease, patients with a history of chronic corticosteroid or anti-inflammatory drug use, pregnant women, or patients who had given birth or had been breastfeeding in the last 90 days, and those who were determined to undergo to an emergency or elective coronary artery bypass surgery (CABG) after angiography, and admission to the hospital after 12:00 PM, those who had claustrophobia or contraindications in cMRI imaging, those who withdrew their consent to participate, those who died in hospital, and those who were dropped out of follow-up or did not experience the second session of cMRI imaging (for non-contact or logistical reasons, rejected, re-infarction, cardiac death or urgent coronary revascularization) were excluded.
The patients were divided into two groups: smokers and non-smokers. In addition, at the 6-month follow-up after STEMI, they were divided into groups: those who developed AR and those who did not.
Study protocol
Clinical, demographic, laboratory and radiological findings were recorded in the patient files during MI. Baseline demographics parameters were age, gender, smoking, history of hypertension, and diabetes mellitus. Baseline clinical parameters were heart rate, systolic and diastolic blood pressure, symptom-to-balloon time, door to balloon time. Baseline laboratory parameters were cardiac troponin I (cTn-I), leukocytes and their subtypes, lipid parameters, hemoglobin, glucose, high sensitive C reactive protein (hs-CRP), and cytokine parameters including interferon (IFN)-α, IFN-γ, IFN-β, interleukin (IL)-6R-α, soluble tumor necrosis factor receptor-1 (sTNFR-1), and sTNFR-2. Radiological parameters were left ventricular (LV) ejection fraction (LVEF), LV end-systolic volume (LVESV), LV end-diastolic volume (LVEDV), cardiac output (CO), and infarct size.
After inclusion, a follow-up cMRI imaging was executed at baseline (2 weeks) and six months after the index event. The inflammatory markers were assessed on the first day (baseline) and two weeks (14 days) after the index event. Serum and plasma were kept at −80°C until assayed. After collecting the serum specimens of the whole sample, parameters were measured by the same device, in the same session, and by the same laboratory staff in the Tissue Typing Laboratory and Genetic Diagnosis Center of the Hospital.
Laboratory parameters
For the measurement of complete blood count and lipid and cardiac biomarkers, venous blood specimens were taken at first admission and centrifuged at 1500 rpm for 10 minutes. Complete blood count parameters were analyzed on the Sysmex XN-1000 (Sysmex Corporation, Kobe, Japan) hematology analyzer, while hemoglobin was measured photometrically. Total cholesterol was measured by the homogeneous enzymatic colorimetric method with a Hitachi Modular P800 autoanalyzer (Roche Diagnostics Corp., Indianapolis, IN, USA). Low-density lipoprotein (LDL) was calculated with Friedewald method (19).
Serum samples were interpreted by the manufacturer's instructions for the bead-based multiplex immunoassay system (Bio-Plex Pro Human Inflammation Panel 1, 37-Plex). Cytokine quantification was conducted in duplicate. Serum specimens were thawed on ice. Bio-Plex MAGPIX System (Bio-Rad, Hercules, CA, USA) was used to measure and quantify the formation of different sandwich immunocomplexes on distinct bead sets. The Bio-Plex Manager v.5.0 software package (Bio-Rad) was used to calculate the final concentration of the analytes. For all statistical analyses, values below the detection limit of the assay were replaced with the minimal detectable value for the analyte. The coefficient of variation between tests for all inflammatory markers were < 15%.
Cardiac magnetic resonance imaging (cMRI)
All cMRI studies were conducted with a 3-T scanner (Magnetom Skyra, Siemens Medical Systems, Erlangen, Germany). The images had a protocol that included access to 4- and 2-chamber views and cine short-axis sections from the bottom to the apex of the heart (slice thickness 6 mm at 10-mm intervals). The indices of LV systolic function were assessed by using a retrospective electrocardiogram-gated turbo-fast low angle shot (turbo-FLASH) sequence with the following setting: echo time (TE) 1.42 msec, repetition time (TR) 39 msec, flip angle 57°, voxel size 1.67 × 1.67 x 6 mm. The syngo via imaging software (Siemens) was used to measure cardiac function and volumes. Thus, LVEDV and LVESV were performed by short-axis-based planimetry from basal to apical level. Stroke volume was computed as LVEDV minus LVESV, and LVEF was calculated as follows: EF = [(LVEDV–LVESV)/LVEDV] x 100.
At 6-month follow-up after STEMI, an increase in LVEDV of 10% or more was considered AR (20).
Statistical analysis
Statistical analysis was conducted utilizing IBM SPSS Statistics for Windows 20.0 (IBM Corp., Armonk, NY, USA). The average allocation of the information was assessed utilizing the Kolmogorov-Smirnov test. Numeric variables with and without regular distribution are presented as the mean (standard deviation) and median [interquartile range (IQR): 25th-75th percentile], respectively. Categorical variables are indicated as the number and percentage. In comparisons between groups, Student`s t test or Mann-Whitney U test were used for continuous variables, while Chi-square, Yates correction and Fischer tests were used for categorical variables. A composite model for replicated measurements (MMRM) analysis was executed to compare the cytokine levels post-MI and follow-up. Independent predictors of AR were evaluated by multiple logistic regression analysis. Consequently, the dependent variable was AR development. Independent variables were defined as those with p-values below 0.25 from the demographic, clinical, and laboratory variables defined in the study protocol title. These potential risk factors were included in the multiple logistic regression models (21). The detection of risk factors impacting the inflammatory marker, which has a significant effect on AR, was examined by robust regression analysis (22, 23). Accordingly, the dependent variable was IFN-β. Independent variables were demographic, clinical, and laboratory variables. P < 0.05 (*) was accepted as statistically significant.
Results
Demographic , clinical, laboratory, echocardiographic, angiographic and cMRI findings of the study population are detailed in Tables 1 and 2. The mean age at admission was 55.4 (6.9) years, consisted mostly of males (n=37), and a hypertension rate of 34.9% with a representative risk profile for cardiovascular disease. At six months post-MI, twenty patients (46.5%) exhibited AR. The smoking ratio was higher in AR group compared to the without AR group (65% vs. 30.4%; p= 0.024) (Tables 1).
Smokers did not differ from non-smokers by demographic, clinical, echocardiographic and angiographic variables and treatment. However, smokers had significantly higher levels of cTn-I (p=0.031), LDL (p=0.003) and hs-CRP (p=0.045) levels as compared to non-smokers.
Patients with AR were more often smokers (p=0.024), had higher cTn-I (p=0.028), neutrophil count (p=0.026) and higher hsCRP (p=0.027) level as compared to patients without AR. There were no differences in other variables between AR groups (Table 1).
|
Table 1. Clinical characteristics of patients with STEMI |
||||||||||
|
Variables |
All population |
Smoking |
p |
Adverse remodeling |
p |
|||||
|
Yes |
No |
Yes |
No |
|||||||
|
n=43 |
n = 20 |
n = 23 |
n = 20 |
n = 23 |
||||||
|
Demographic findings |
||||||||||
|
Age, years |
55.4(6.9) |
54.4(6.3) |
56.3(7.5) |
0.387 |
54.5(6.2) |
56.2(7.5) |
0.437 |
|||
|
Male gender, n(%) |
37(86.0) |
16(80.0) |
21(91.3) |
0.531 |
17(85.0) |
20(87.0) |
0.999 |
|||
|
Smoking, n(%) |
20(46.5) |
20(100) |
23(100) |
- |
13(65.0) |
7(30.4) |
0.024* |
|||
|
Hypertension, n(%) |
15(34.9) |
7(35.0) |
8(34.8) |
0.999 |
9(45.0) |
6(26.1) |
0.219 |
|||
|
Diabetes mellitus, n(%) |
10(23.3) |
4(23.3) |
6(26.1) |
0.913 |
6(30.0) |
4(17.4) |
0.539 |
|||
|
Clinical findings |
||||||||||
|
Heart rate, bpm |
72.2(12.0) |
75.3(11.5) |
69.6(12.1) |
0.127 |
73.8(7.8) |
70.9(14.8) |
0.430 |
|||
|
SBP, mm Hg |
122.1(14.2) |
121.1(13.5) |
123.0(15.1) |
0.676 |
121.0(9.5) |
123.1(17.5) |
0.617 |
|||
|
DBP, mm Hg |
77.8(12.8) |
75.1(10.0_ |
78.3(15.0) |
0.420 |
77.5(9.5) |
78.0(15.4) |
0.882 |
|||
|
Echocardiographic findings |
||||||||||
|
LVEF, % |
46.4(8.5) |
45.8(7.8) |
47.0(8.6) |
0.636 |
45.4(8.1) |
47.2(8.8) |
0.492 |
|||
|
LV EDV, mL |
154.8(30.5) |
150.1(30.9) |
158.1(33.2) |
0.421 |
149.1(23.2) |
159.3(31.6) |
0.241 |
|||
|
LV ESV, mL |
80(55-128) |
72(50-112) |
88(55-130) |
0.251 |
90(64-132) |
70(50-124) |
0.878 |
|||
|
Coronary angiographic findings |
||||||||||
|
DBT, min |
42.8(8.3) |
42.2(8.1) |
43.1(8.5) |
0.373 |
43.4(8.4) |
42.3(8.6) |
0.675 |
|||
|
SBT, min |
316.5(68.5) |
310.7(59.0) |
319.4(75.0) |
0.483 |
313.4(66.2) |
318.8(71.5) |
0.800 |
|||
|
Culprit lesion, n(%) |
|
|||||||||
|
LAD |
29(67.4) |
16(80.0) |
13(56.5) |
0.119 |
14(70.0) |
15(65.2) |
0.994 |
|||
|
Cx |
14(32.6) |
4(20.0) |
10(43.5) |
6(70.0) |
8(34.8) |
|||||
|
Laboratory findings |
||||||||||
|
cTn-I, µg/L |
47(35.3-59.7) |
55.3(43.2-64.3) |
40.6(30.249.5) |
0.031* |
58.4(8.9-31.2) |
41.3(10.3-29.5) |
0.028* |
|||
|
WBC, x109/ L |
11.4(2.7) |
12.1(2.8) |
10.8(2.4) |
0.116 |
11.6(2.8) |
11.2(2.6) |
0.682 |
|||
|
Neutrophil, x109/ L |
8.7(1.9) |
9.2(1.8) |
8.3(2.0) |
0.129 |
9.4(1.5) |
8.1(2.1) |
0.026* |
|||
|
Lymphocyte, x109/ L |
2.3(0.7) |
2.2(0.8) |
2.3(0.6) |
0.911 |
2.1(0.6) |
2.4(0.7) |
0.058 |
|||
|
Monocyte, x109/ L |
0.8(0.1) |
0.8(0.1) |
0.8(0.2) |
0.560 |
0.8(0.1 |
0.8(0.2) |
0.705 |
|||
|
Platelet, x109/ L |
331.4(37.4) |
338.6(36.6) |
325.1(37.6) |
0.243 |
341.9(34.8 |
322.2(37.8) |
0.084 |
|||
|
Hemoglobin, g/dL |
13.8(2.2) |
13.8(1.9) |
13.8(2.4) |
0.92 |
13.9(2.2 |
13.8(2.2) |
0.885 |
|||
|
Glucose, mg/dL |
105.2(22.6) |
108.5(19.5) |
102.3(25) |
0.379 |
108.9(23.6 |
102.0(21.7) |
0.324 |
|||
|
HDL, mg/dL |
39.5 (10.7) |
39.6 (9.6) |
39.4 ( 11.9 |
0.961 |
39.2(10.6 |
39.8(11.1) |
0.862 |
|||
|
LDL, mg/dL |
104(70-136) |
133.5(106-153) |
98(67-121) |
0.003* |
108(83-144.5) |
104(67-133) |
0.519 |
|||
|
hs-CRP, mg/L |
24.7(16-33.6) |
39.8(26.5-45.9) |
20.6(14-33) |
0.045* |
28.7(20.8-42.2) |
20.1(11-26.8) |
0.027* |
|||
|
Discharge therapy, n (%) |
||||||||||
|
ACE/ARB, |
43(100) |
20(100) |
23(100) |
- |
20(100) |
23(100) |
- |
|||
|
Beta blockers |
40(93.0) |
18(90.0) |
22(95.7) |
0.900 |
19(95.0) |
21(91.3) |
0.999 |
|||
|
Statins |
38(88.4) |
19(95.0) |
19(82.6) |
0.431 |
18(90.0) |
20(87.0) |
0.999 |
|||
|
Categorical variables are shown as n(%). Continuous variables with normal distribution are shown as mean (SD), while those with abnormal distribution are shown as median (IQR). * p <0.05 shows statistical significance cTn-I - cardiac troponin I, CO - cardiac output, DBP - diastolic blood pressure, DBT - door- to- balloon time, EDV - end-diastolic volume, EF - ejection fraction, ESV - end-systolic volume, HDL - high density lipoprotein, hs-CRP - high sensitive C- reactive protein, IS- infarct size; LDL- low density lipoprotein, LV - left ventricular, SBP - systolic blood pressure, SBT - symptom- to- balloon time, STEMI – ST-elevation myocardial infarction, WBC - white blood cell |
||||||||||
cMRI data are presented in Table 2. cMRI analysis demonstrated that smokers had AR twice higher than non-smokers (p=0.024). Patients with AR had larger infarct size (p=0.038) 2 weeks after STEMI as compared to patients without AR. At 6-month they had significantly lower LV EF (p=0.014), larger volumes (LVEDV –p<0.001 and LVESV –p=0.001) and extensive infract size (p=0.012) as compared to patients without AR.
|
Table 2. cMRI findings of patients with STEMI |
|||||||
|
Variables |
All Population
|
Smoking
|
p |
Adverse remodeling |
p |
||
|
n=43 |
Yes |
No |
Yes |
No |
|||
|
n = 20 |
n = 23 |
n = 20 |
N=23 |
||||
|
2 weeks |
|||||||
|
LV EF, % |
47.2(10.6) |
47.9(11.2) |
46.6(10.3) |
0.686 |
47.9(10.7) |
46.6(10.8) |
0.707 |
|
LV EDV, mL |
160.4(33.8) |
155.7(32.6) |
164.5(35) |
0.399 |
158.4(27.6) |
162.1(38.9) |
0.726 |
|
LV ESV, mL |
84(59-133) |
70(55.5-116.5) |
93(61-144) |
0.176 |
99(57.5-130.5) |
72(60-144) |
0.827 |
|
CO, mL/min |
4.7(1.1) |
4.7(1.0) |
4.6(1.1) |
0.801 |
4.8(1.3) |
4.6(0.8) |
0.541 |
|
IS, % of LV |
17(12-23) |
19.5(14-27) |
15(11-22) |
0.192 |
20(14.5-26) |
15(10-22) |
0.038* |
|
6 months |
|||||||
|
LV EF, % |
47.7(10.4) |
47.1(11.1) |
48.3(10) |
0.699 |
43.6(10.7) |
51.3(9.0) |
0.014* |
|
LV EDV, mL |
163.7(38.6) |
170.1(38.8) |
158.1(38.4) |
0.317 |
186.4(32.3) |
143.9(32.7) |
<0.001* |
|
LV ESV, mL |
79(58-132) |
76(60-131) |
79(58-134) |
0.961 |
119.5(71-159) |
62(53-121) |
0.001* |
|
CO, mL/min |
4.7(1.2) |
4.5(1.1) |
4.9(1.3) |
0.336 |
4.6(1.6) |
4.8(0.8) |
0.687 |
|
IS, % of LV |
11(8-16) |
13(9-21) |
10(8-15) |
0.205 |
15(10-20) |
10(7-14) |
0.012* |
|
AR, n (%) |
20(46.5) |
13(65.0) |
7(30.4) |
0.024* |
20(100) |
23(100) |
- |
|
Categorical variables are shown as n(%). Continuous variables with normal distribution are shown as mean (SD), while those with abnormal distribution are shown as median (IQR). * p <0.05 shows statistical significance cMRI – cardiac magnetic resonance imaging, CO - cardiac output, EDV - end-diastolic volume, EF - ejection fraction, ESV - end-systolic volume, IS- infarct size, LV - left ventricular, STEMI – ST-elevation myocardial infarction |
|||||||
The median IFN-α (51.7 vs. 44.3 pg/mL; p= 0.001, respectively), median IFN-γ levels (21.7 vs. 16.2 pg/mL; p< 0.001), median IFN-β (38.5 vs. 23 pg/mL; p< 0.001, respectively), median IL-6R-α (19051.6 vs. 12262.3 pg/mL; p< 0.001, respectively), median sTNFR-2 (6205.3 vs. 3585.2 pg/mL; p< 0.001, respectively) baseline concentrations were higher in smokers group compared to non-smokers group. At the 2 weeks post-MI, cytokine concentrations were similar in smokers and non-smokers (p> 0.05) (Table 3).
In AR group compared to without AR group, the median IFN-α (48.7 vs. 44.5 pg/mL; p= 0.015, respectively), median IFN-β (33.8 vs. 23.2 pg/mL; p= 0.008, respectively), median IL-6R-α (15884.6 vs. 13104 pg/mL; p= 0.036, respectively) baseline concentrations were higher. At the two weeks post-MI, cytokine concentrations were similar in AR group compared to the without AR group (p> 0.05) (Table 3).
In the non-smokers' group, cytokine concentrations on the first day and two weeks post-MI were similar in AR group compared to the without AR group. In the smokers' group, median IFN-α (54.5 vs. 47.2 pg/mL; p= 0.024, respectively) and IFN-β (44.5 vs. 31.1 pg/mL; p= 0.014, respectively) baseline concentrations were higher in AR group compared to without AR group. Other cytokine concentrations were not remarkably different in patients with and without AR (Table 4).
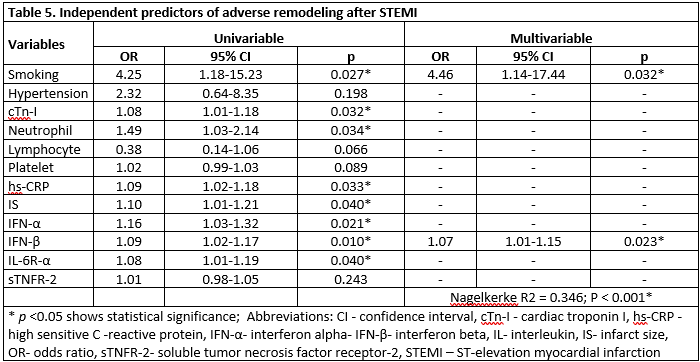
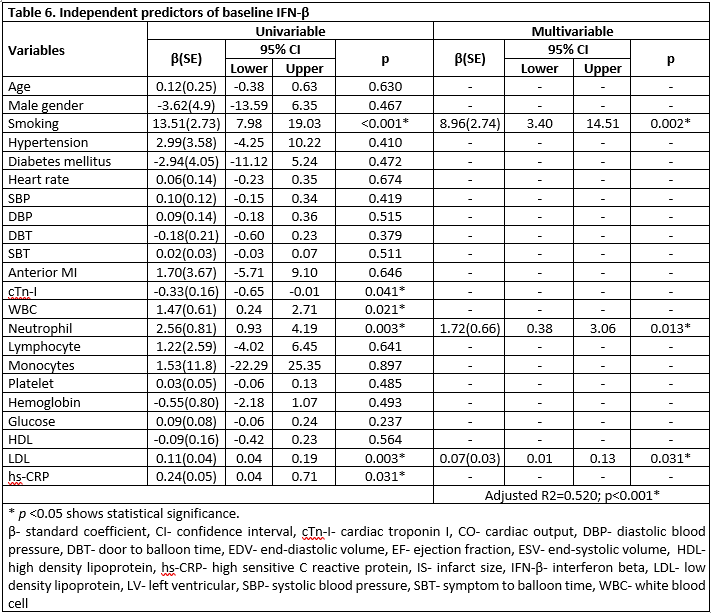
In the multiple logistic regression analysis, smoking (OR= 4.46; p= 0.032) and IFN-β (OR= 1.07; p= 0.023) were independent predictors of AR (Table 5). Furthermore, smoking (β(SE)= 8.96(2.74); p= 0.002) neutrophil levels (β(SE)= 1.72(0.66); p= 0.013) and LDL levels (β(SE)= 0.07(0.03); p= 0.031) were independent predictors of elevated IFN-β concentration (Table 6). The relationship between IFN-β and LDL is shown in Figure 1.
|
Table 3. Distribution of cytokine levels in smoking and adverse remodeling |
|||||||
|
Cytokines |
Smoking |
p |
Adverse remodeling |
p |
|||
|
Yes |
No |
Yes |
No |
||||
|
n = 20 |
n = 23 |
n = 20 |
n = 23 |
||||
|
IFN-α, pq/mL |
|||||||
|
First day |
51.7 (45.8-55.4) |
44.3 (41.5-47.2) |
0.001* |
48.7 (44.8-55.4) |
44.5 (41.5-50.1) |
0.015* |
|
|
2 weeks |
36.6 (28.6-44.3) |
38.6 (30.4-47.2) |
0.556 |
37.2 (28.6-42.9) |
44.1 (30.4-50.1) |
0.143 |
|
|
IFN-γ, pq/mL |
|||||||
|
First day |
21.7 (20.4-26.1) |
16.2 (10.3-16.2) |
<0.001* |
20.4 (16.2-24.2) |
16.5 (13.3-21.7) |
0.259 |
|
|
2 weeks |
26.1 (17.7-31) |
24.2 (16.2-32.2) |
0.494 |
23.3 (15.5-31.0) |
24.9 (19.0-32.2) |
0.502 |
|
|
IFN-β, pq/mL |
|||||||
|
First day |
38.5 (29.8-44.5) |
23.0 (17.6-25.7) |
<0.001* |
33.8 (24.4-44.5) |
23.2 (17.6-31.1) |
0.008* |
|
|
2 weeks |
31.1 (25.3-36.5) |
28.4 (23.0-32.3) |
0.150 |
27.6 (23.0-35.1) |
31.1 (25.7-33.8) |
0.696 |
|
|
IL-6R-α, pq/mL |
|||||||
|
First day |
19051.6 (15735.8-25903.5) |
12262.3 (9561.7-14103.6) |
<0.001* |
15884.6 (13568.4-21554.4) |
13104.0 (10688.5-17024.5) |
0.036* |
|
|
2 weeks |
15555 (14108.7-18387) |
13128.0 (10630.6-18476.9) |
0.173 |
14668.9 (10685.3-17776.6) |
14896.3 (12538.6-19904.5) |
0.480 |
|
|
sTNFR-1, pq/mL |
|||||||
|
First day |
2694.5 (2108.6-3407.6) |
2125.8 (1668.8-2957.7) |
0.061 |
2643.4 (2105.7-3144.2) |
2300 (1683.5-3427.6) |
0.465 |
|
|
2 weeks |
2958.2 (2211.5-3469.7) |
2499.9 (1704.9-2912) |
0.054 |
2630.9 (1925.4-3276.2) |
2785 (1791.2-3031.7) |
0.752 |
|
|
sTNFR-2, pq/mL |
|||||||
|
First day |
6205.3 (5085.3-8166) |
3585.2 (2083-4792.7) |
<0.001* |
5101 (4018.6-7726.5) |
4072.2 (2642.8-6172.5) |
0.165 |
|
|
2 weeks |
6253.6 (4343-8739.3) |
4875.4 (4077-6293.3) |
0.113 |
5148 (4129.9-7510.7) |
5709.5 (4289.2-6937.9) |
0.865 |
|
|
Cytokines levels are shown as the median (IQR). * p <0.05 shows statistical significance IFN-α - interferon alpha, IFN-γ - interferon gamma; IFN-β - interferon beta, IL - interleukin, sTNFR-1 - soluble tumor necrosis factor receptor-1, sTNFR-2 - soluble tumor necrosis factor receptor-2 |
|||||||
Discussion
The main results of this study are: 1) the ratio of smokers was higher in the patient with AR. 2) The concentrations of IFN-α, IFN-γ, IFN-β, IL-6R-α, and sTNFR-2 on the first-day post-MI were higher in smokers. 3) In AR, the concentrations of IFN-α, IFN-β, and IL-6R-α on the first-day post-MI were higher. 4) Smoking and high IFN-β baseline concentration were independent predictors of AR. 5) Smoking, increased neutrophil, and increased LDL were independent predictors of elevated IFN-β.
It is known that smokers have a high risk of cardiovascular disease, including MI. A single cigarette contains more than 4000 chemical components, which has quite a higher number of potential adverse effects on cardiac tissue (24). Rafacho et al. (1) found that the stimulated activation of NADPH oxidase with exposure to smoking in rats caused the development of ventricular dysfunction. A study on rats conducted by Bradley et al. (24) indicated that chronic exposure to smoking results in abnormal dilatation and impaired cardiac function by stimulating matrix metalloproteinase-9 and matrix metalloproteinase tissue inhibitor expression in cardiac tissue with volume overload. A retrospective study in humans with coronary computed tomography angiography identified smoking as an independent risk factor for arterial expansion (25). Oakes et al. (26) investigated the relationship between chronic nicotine inhalation in rats with systemic and pulmonary hypertension and ventricular remodeling. They found that chronic inhalation increased systemic and pulmonary blood pressure and proper ventricular AR, possibly causing progressive and permanent pulmonary hypertension (26).
|
Table 4. Distribution of cytokine levels in with and without AR groups by smoking status |
||||||
|
Cytokines |
Non-smokers |
p |
Smokers |
p |
||
|
AR group (n = 7) |
without AR group (n = 16) |
AR group (n = 13) |
without AR group (n = 7) |
|||
|
IFN-α, pq/mL |
||||||
|
First day |
45.3 (41.5-47.2) |
42.9 (40.1-47.2) |
0.535 |
54.5 (50.1-55.9) |
47.2 (41.5-53) |
0.024* |
|
2 weeks |
30.4 (23.2-38.6) |
44.3 (30.4-48.7) |
0.089 |
35.7 (30.4-44.3) |
44.3 (26.8-50.1) |
0.438 |
|
IFN-γ, pq/mL |
||||||
|
First day |
16.2 (10.3-16.2) |
14.8 (11.1-17.6) |
0.820 |
21.9 (19-26.7) |
21.7 (21.7-25.5) |
0.817 |
|
2 weeks |
16.4 (6.9-29.1) |
24.2 (19.0-32.2) |
0.413 |
24.8 (16.2-32.2) |
27.3 (21.7-29.1) |
0.699 |
|
IFN-β, pq/mL |
||||||
|
First day |
23 (14.8-28.4) |
23.0 (19.0-25.7) |
0.769 |
44.5 (39.1-47.1) |
31.1 (17.6-36.5) |
0.014* |
|
2 weeks |
23.9 (21.1-31.1) |
28.4 (24.4-33.1) |
0.249 |
31.1 (23-43.3) |
30.1 (29.5-33.8) |
0.938 |
|
IL-6R-α, pq/mL |
||||||
|
First day |
13033.1 (11490.5-14772) |
11889.4 (9133.5-13206) |
0.341 |
18753.3 (16209.2-23834.3) |
19349.9 (14485.7-27972.6) |
0.817 |
|
2 weeks |
12004.5 (10198.9-17448.9) |
13829.3 (11336-19190.7) |
0.671 |
14951.5 (13407.3-17886.7) |
16158.5 (14842.9-31313.5) |
0.183 |
|
sTNFR-1, pq/mL |
||||||
|
First day |
2702.3 (1967.9-3123) |
1906 (1639.9-2919.7) |
0.308 |
2595.6 (2131.6-3165.4) |
3310.4 (1996.5-3890.7) |
0.351 |
|
2 weeks |
2607.3 (1716.2-2881.7) |
2420.1 (1672.4-2928) |
0.830 |
2840.8 (2213.1-3447.1) |
3106.4 (2209.8-3536.2) |
0.643 |
|
sTNFR-2, pq/mL |
||||||
|
First day |
3550.3 (2053.9-4776.2) |
3644 (2210.3-5122.2) |
0.769 |
7030.5 (5409.2-8136.5) |
6172.5 (4072.2-9992.5) |
0.938 |
|
2 weeks |
4593.8 (3634-5363.9) |
5636.8 (4313.7-6434.6) |
0.175 |
5400.2 (4577.9-8423.1) |
7317.5 (4214.1-9055.4) |
0.877 |
|
Cytokines levels are shown as the median (IQR)., * p <0.05 shows statistical significance. Abbreviations: AR - adverse remodeling, IFN-α - interferon alpha, IFN-γ - interferon gamma, IFN-β - interferon beta, IL - interleukin, sTNFR-1 - soluble tumor necrosis factor receptor-1, sTNFR-2 - soluble tumor necrosis factor receptor-2 |
||||||
Wu et al. (27) studied the role of passive smoking in old rats. Passive exposure to smoking increased LV fibrosis in rats, and the elevation of matrix metalloproteinase levels by smoking was considered the potential mechanism (27).
In smokers, the fate of prognosis post-MI, including ventricular remodeling, depends on a complex mechanism, including the direct effect of smoking by triggering left atrial and ventricular dilatation, causing myocyte hypertrophy and LV dysfunction (28, 29). Cigarette smoking leads to an excessive inflammatory response (30), stimulation of reactive oxygen radical levels (1), the onset of atherothrombosis and its progression via lipid modification (31), and endothelial dysfunction resulting in elevated blood pressure and impaired cardiac structure and function (5). The basis of these mechanisms is the inflammatory response. Therefore, we evaluated some of the inflammatory markers, including IFN-α, IFN-γ, IFN-β, IL-6R-α, sTNFR-1, and sTNFR-2. The current study indicated smoking and IFN-β were independent risk factors in AR. Furthermore, on the first-day post-MI, higher inflammatory markers levels in smokers suggest that smoking plays a role in the stimulation of inflammation in AR. Our study shows that smoking could play a role in increased IFN-β levels post-MI. Haig et al. (32) found higher C-reactive protein levels, neutrophil, and monocyte counts in smokers compared to non-smokers among a population of 324 STEMI patients and found an independent relationship between smoking and systemic inflammation. In light of these findings, smokers may be at risk of AR in the six months due to hyper-inflammation at the onset of MI. Thus, the risk of heart failure can become inevitable in the long-term.
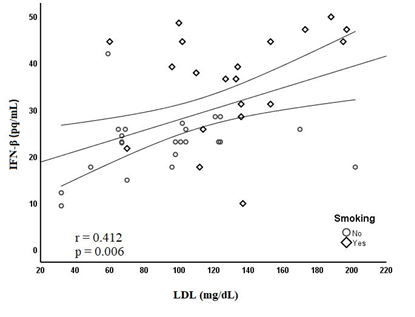
Figure 1. Relationship between IFN-β and LDL
IFN-β - interferon beta, LDL - low-density lipoprotein
Although the development of AR after MI is associated with an increased risk of mortality (33-35), there are reports to the contrary (36). However, AR is known to cause heart failure, functional mitral regurgitation and ventricular arrhythmias (36, 37). Therefore, it may have a direct or indirect effect on mortality. Moreover, overproduction of type I interferons at the time of MI has been shown to trigger a lethal response (38). On the other hand, higher admission CRP levels, a marker of increased inflammation, in acute heart failure were associated with higher mortality (39, 40). The current study showed that smoking is associated with higher inflammation and higher levels of type I interferons and plays a role in the development of AR.
LDL levels were higher in smokers and were an independent predictor of INF-β. The relation of smoking and LDL cholesterol with inflammation can be linked to compounds, frequently reactive oxygen radicals, causing inflammation via activation of epithelial intracellular signal cascades resulting in inflammatory gene activation (30, 41). LDL cholesterol activates toll-like receptor pathways triggering pro-inflammatory signals and stimulates innate immune system activation, which results in increased active pro-inflammatory cytokine levels (42). Although statin therapy is effective in reducing LDL levels, only a small proportion of patients achieve their LDL-cholesterol goals (43). Statins have pleiotropic effects such as anti-inflammatory and antithrombotic properties and antioxidant effects. Thus, it plays a role in reducing the cellular inflammatory response after STEMI (44). A meta-analysis study suggested that statin have light efficacy in treating or reducing the risk of MI patients (45). On the other hand, in an experimental study, statins were shown to improve cardiac remodeling by stimulating new myocyte formation after MI (46). It is known that a significant proportion of MI patients treated in accordance with the guidelines are at risk of AR (47). In this study, all patients were treated according to the guidelines and no significant association was found between discharge treatments, including statins, and the development of AR.
Pentoxifylline may effectively alleviate cardiac remodeling post-MI in smokers patients (4, 48). Sliwa et al. (49) added pentoxifylline to the treatment regimens of 38 ischemic cardiomyopathy patients, an immunomodulatory molecule and a suppressant of TNF-α. The decreased plasma inflammatory marker levels improved clinical status and radionuclide ejection fraction. Fernandes et al. (50) studied pentoxifylline in 68 patients diagnosed with the acute coronary syndrome. They randomized these patients into two groups, receiving either a placebo or pentoxifylline. The group receiving the pentoxifylline treatment, a phosphodiesterase inhibitor, had decreased pro-inflammatory cytokine levels, such as TNF-α, and increased anti-inflammatory cytokine levels, such as IL-10.
Study limitations
The major limitation of the current study was its sectional design, which prevented definite conclusions from being drawn regarding the effect of smoking on increased inflammation in AR. In addition, we could not evaluate the number of cigarettes smoked daily by the patients. This may affect cardiac function due to the accumulation of nicotine and tar in smokers. Another limitation was the relatively low number of patients in the study.
Conclusion
Smokers had higher inflammation levels at the onset of acute MI. In addition, a higher incidence of AR was observed after MI in these patients. Smoking may contribute to the development of AR by increasing the severity of inflammation at the onset of acute MI.
Ethics: The study was performed in accordance with the Declaration of Helsinki (revised 2013) and was approved by the Ankara Yildirim Beyazit University Faculty of Medicine Non-Drug Clinical Research Ethics Committee on 24 June 2013 under Decision No. 2013/106. Written informed consent was obtained from all patients.
Peer-review: External and internal
Conflict of interest: None to declare
Authorship: Concept & Design – C.S.; Data collection and/or processing – C.S., N.E., O.K., M.A.F., E.K., and O.F.A.; Analysis and/or interpretation – C.S., N.E., O.K., M.A.F., E.K., and O.F.A.; Literature search – C.S., N.E., O.K., M.A.F., E.K., and O.F.A.; Writing – C.S.; Critical review – N.E. and O.F.A. All authors have read and approved the final version of the study.
Acknowledgement : None
Funding: The authors declare that this research received financial support from the Ministry of Health of the Republic of Turkey (Department of Research, Development, and Health Technology Evaluation) through project 2015/SAGEM-2/001.
References
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Copyright

This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.
AUTHOR'S CORNER
