Case report of symptomatic very late presentation of ALCAPA syndrome: from AL-CAPONE to Robin Hood of coronary artery anomalies
CASE REPORT
Case report of symptomatic very late presentation of ALCAPA syndrome: from AL-CAPONE to Robin Hood of coronary artery anomalies
Article Summary
- DOI: 10.24969/hvt.2023.394
- CARDIOVASCULAR DISEASES
- Published: 05/06/2023
- Received: 23/02/2023
- Revised: 23/05/2023
- Accepted: 25/05/2023
- Views: 6645
- Downloads: 4863
- Keywords: Bland-White-Garland syndrome, myocardial infarction, congenital diseases, ALCAPA
Address for Correspondence*: Raul Cruz Palomera*, Instituto Mexicano Del Seguro Social. 2 Norte 2004 Col Centro, Puebla, Puebla, Mexico Phone: 222 242 4520 Email: Raulcp777@gmail.com ORCID - https://orcid.org/0000-0002-1347-7858 Twitter - @raulcp777_raul
Raul Cruz Palomera*, Rosa Elena Gutierrez Castañeda , Juan Francisco Rodriguez Alvarado, Juan Guzman Olea, Gabriel Guzman Olea, Jorge Guillermo Arenas Fonseca, Zuriel Almeyda Dominguez, Rolando Vicente Colmenares
Instituto Mexicano Del Seguro Social, Puebla, Mexico
Abstract
Objective: Anomalous origin of the left coronary artery from the pulmonary artery (ALCAPA), also known as Bland-White-Garland syndrome, is a rare congenital condition, which can manifest as various cardiac symptoms.
Case presentation: A 64-year-old male patient presented with functional class deterioration and typical angina to hospital. He was stratified by an ischemia-induced study and underwent coronary angiography that found to have ALCAPA.
Conclusions: This is a rare case of ALCAPA due to the patient´s age. Survival to adulthood is possible due to collateral circulation, though the cost is the presence of heart failure, and wherein due to the high surgical risk, in this case optimal medical therapy was decided.
Key words: Bland-White-Garland syndrome, myocardial infarction, congenital diseases, ALCAPA
Abbreviations:
ALCAPA - anomalous origin of the left coronary artery from the pulmonary artery
ICD - implantable cardioverter-defibrillator
LVEF - left ventricular ejection fraction
RCA - right coronary artery
Introduction
The anomalous origin of the left coronary artery from the pulmonary artery “ALCAPA” or Bland-White-Garland syndrome has been reported with an incidence of 0.25 to 0.5% of all isolated congenital heart disease (1, 2). It has been related to other congenital heart diseases such as atrial and interventricular septal defects, patent ductus arteriosus, aortic coarctation or other anomalies of the aortic arch, and tetralogy of Fallot (3).
Mortality is very high, 90% in the first year of life, if corrective surgery is not performed. In cases where there is adequate collateral circulation between the right coronary artery to the left coronary system, they can survive beyond the first year of life (adult type) (1, 4).
Case report
A 64-year-old man without modifiable cardiovascular risk factors, with a cardiovascular history of atrial fibrillation and heart failure was admitted to the hospital. He had started experiencing the symptomatology 6 months ago with dyspnea (NYHA II) and typical angina (CCS II), on treatment with sacubitril/valsartan, spironolactone, metoprolol, dapagliflozin. Physical examination was normal. The electrocardiogram revealed atrial fibrillation rhythm, left anterior fascicular block and QS pattern in V1 to V2 (Fig. 1). Laboratory blood and urine studies were normal. Risk stratification was performed with a stress nuclear study that showed evidence of infarction in the antero-apical wall and moderate inferior ischemia (Fig. 2). Therefore, he underwent coronary angiography that revealed dilated right coronary artery (RCA) originating from the right coronary cusp, and delayed enhanced-imaging demonstrated the left coronary artery (LCA) filling via extensive intercoronary collateral circulation which connects directly to the pulmonary trunk (Fig. 3).
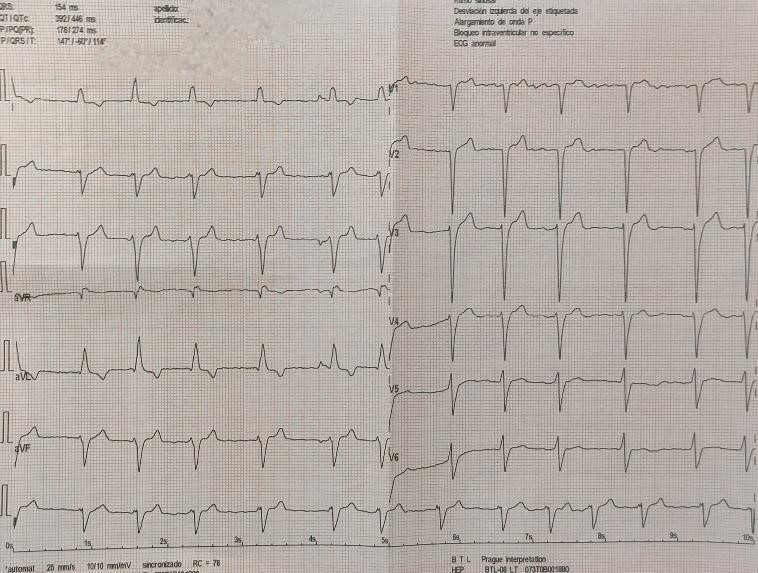
Figure 1. Electrocardiogram demonstrating atrial fibrillation rhythm, left anterior fascicular block and QS pattern in V1 to V2
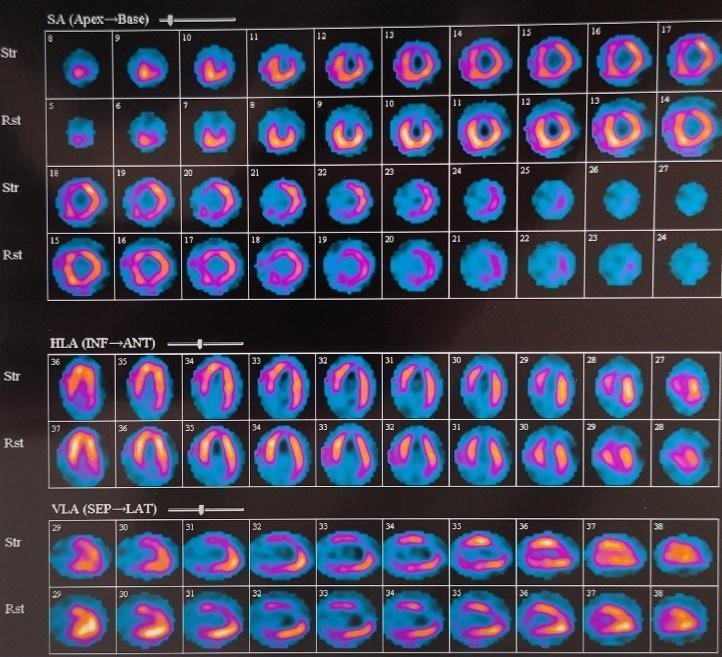
Figure 2. Single-photon emission computed tomography with evidence of infarction in the antero-apical wall and moderate inferior ischemia
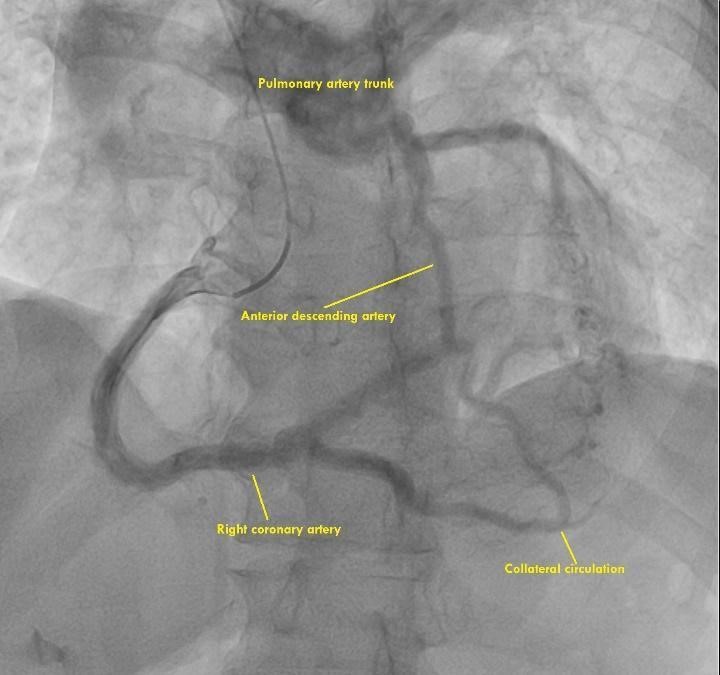
Figure 3. Coronary angiography showing the dilated right coronary artery with collaterals to the left coronary artery
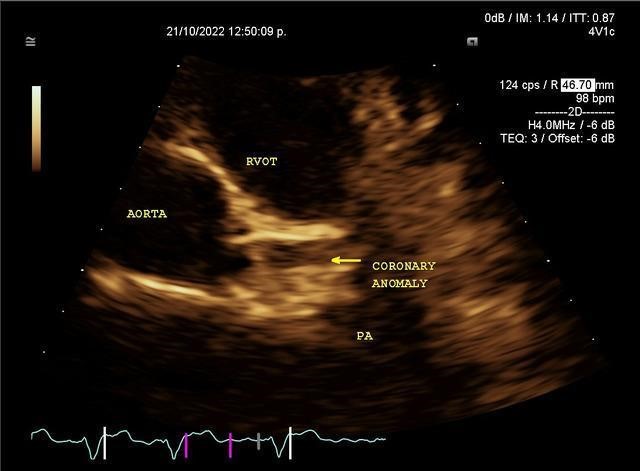
Figure 4. Parasternal short-axis window on transthoracic echocardiography
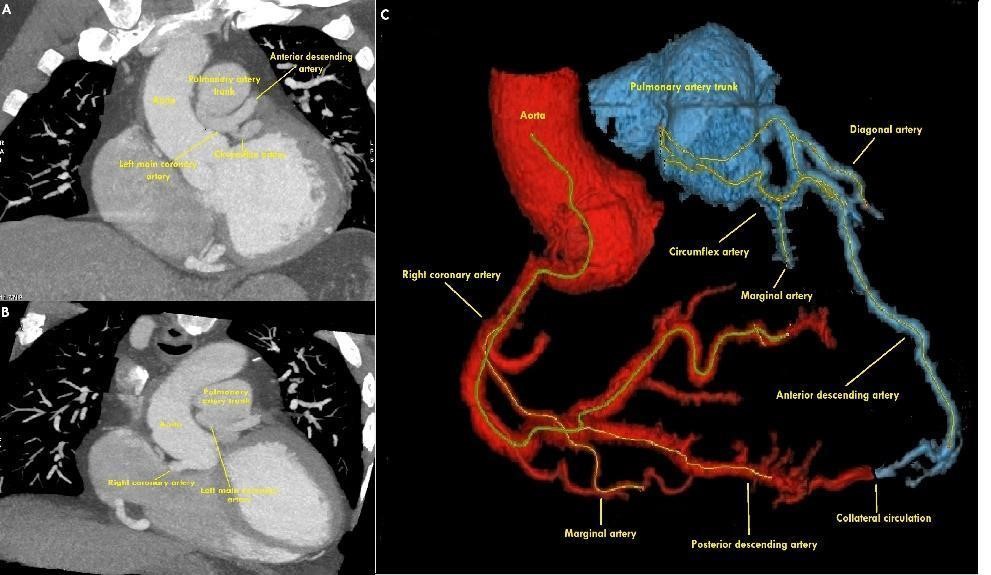
Figure 5. Reconstructed CT image shows the left main coronary artery arising from the pulmonary artery trunk
CT computed tomography
Transthoracic echocardiography with evidence of left ventricular ejection fraction (LVEF) of 35%, moderate mitral regurgitation, dilated left ventricle with apical, anteroseptal, inferoseptal and anterior akinesia, in parasternal short-axis-base view showed origin anomalous of the left main coronary artery from the pulmonary artery trunk (Fig. 4). Computed tomography angiography (CTA) showed the origin of the left system from the lateral wall of the pulmonary artery trunk, without evidence of plaque or stenosis (Fig. 5).
The case was discussed with heart team and it was concluded that due to evidence of an infarction in the territory corresponding to the left system and without myocardial viability, it was decided to continue optimal medical treatment and adequate control of heart failure.
Discussion
Coronary anomaly can be defined as a coronary pattern or characteristic found in less than 1% of the general population. Within the most common types is a separate origin of left anterior descending artery (LAD) and left circumflex artery (LCX), with an incidence of 0.41%, followed by LCX arising from RCA, with an incidence of 0.37%. ALCAPA is a very rare congenital coronary anomaly (0.008%). The presence of ALCAPA in the adult stage is rare, between the ages of 35 and 50 years it has been observed that they can generate symptoms of exertional angina, heart attack, dyspnea, heart failure, syncope or sudden cardiac death (5, 6).
It is well known that in fetal life the pressures of the pulmonary system are elevated with oxygenated blood, therefore in the left system with origin from pulmonary system complications do not exist. After birth the pressures drop, in case of presenting an adequate collateral circulation from the RCA to the left system, it generates a coronary steal phenomenon by the blood that passes from the high-pressure coronary circulation to the lower-pressure pulmonary circulation. A well collateralized and pressurized system would avoid significant hypoperfusion and allow survival into adulthood (5).
The diagnostic methods mentioned in the literature are CTA with reconstruction of the arteries and coronary angiography (5). However, on ECG we can found no obvious ST-T change. Ideally echocardiography should be the initial diagnostic tool, considering this diagnosis when echocardiography shows left ventricular dilation and dysfunction with severe mitral regurgitation, abnormal flow-pattern in the ventricular septum and dilatation of the RCA, wide enhancement and thickening of the endocardium. The typical and direct echocardiographic imaging feature is to show the LCA originating from the pulmonary artery when diagnosing ALCAPA and should also reveal the origin, size, and course of coronary arteries clearly. In addition, the reversal of blood flow in the LCA and abundant collateral circulation are approval to the diagnosis of ALCAPA. Nonetheless, coronary angiography is considered the gold standard in the diagnosis of ALCAPA. Magnetic resonance angiography (MRA) and CTA have been increasingly employed for the diagnosis of ALCAPA (12).
When ALCAPA is diagnosed, even in asymptomatic patients, surgery is suggested for treatment to prevent irreversible myocardial infarctions, malignant ventricular arrhythmias, and sudden cardiac death (1, 7).
Treatment is surgery, for the pediatric population, it consists of reimplantation of the left system in the aorta or bypass graft. If the above techniques are not feasible, due to anatomical limitations or significant comorbidities, closure of the anomalous artery with surgical ligation or percutaneous embolization and revascularization by internal mammary artery graft may result in improvement (8, 9).
Conclusion
This case represents a typical ALCAPA syndrome in adulthood that constitutes the 10% of the patients who survive at one year due to collateral circulation of the RCA. Our patient presented with a functional class deterioration (NYHA II) with LVEF of 35% secondary to an ischemic origin. When making the confirmatory diagnosis, due to the patient's conditions, such as heart failure with reduced left ventricular ejection fraction and high corrected surgical risk a treatment for heart failure was chosen. The optimal treatment of heart failure should be sustained for at least 3 months as mentioned in the ESC/AHA guidelines (10, 11). If the ejection fraction does not improve, primary prevention with an implantable cardioverter-defibrillator (ICD) should beconsidered. So the most important thing in this case is to make a timely diagnosis of it.
Peer-review: External and internal
Conflicts of interest: None to declare
Authorship: R.C.P., R.E.G.C. , J.F.R.A., J.G. O., G.G.O., J.G. A.F., Z.A.D., R.V.C. equally participated in case management and manuscript preparation and fulfilled authorship criteria
Acknowledgement and funding: None to declare
Ethics: Written informed consent was obtained from patient before all procedures
References
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Copyright

This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.
AUTHOR'S CORNER
