Amyotrophic lateral sclerosis in a combination with frontotemporal dementia: A clinical case study
CASE REPORT
Amyotrophic lateral sclerosis in a combination with frontotemporal dementia: A clinical case study
Article Summary
- DOI: 10.24969/hvt.2024.504
- Page(s): 417-420
- CARDIOVASCULAR DISEASES
- Published: 20/08/2024
- Received: 14/03/2024
- Revised: 30/07/2024
- Accepted: 31/08/2024
- Views: 5278
- Downloads: 3907
- Keywords: amyotrophic lateral sclerosis, frontotemporal dementia, atypical form, obesity treatment, eating disorder
Address for Correspondence: Begimai Kadyrova, International School of Medicine of International University of Kyrgyzstan, April 7 Str, 6, 720065, Bishkek, Kyrgyz Republic
E-mail: begimai.kadyrova@gmail.com
ORCID: Nurzhan T. Dzhaparalieva – 0000-0003-0443-2639; Eldana B.Bolotbekova - 0000-0001-5883-3345; Begimai B. Kadyrova - 0000-0003-3208-5689; Irina A. Sverdlova - 0000-0002-3511-242X; Asel T. Jusupova - 0000-0001-8430-9504
Nurzhan T. Dzhaparalieva1, Eldana .B.Bolotbekova1, Begimai B. Kadyrova2, Irina A. Sverdlova1, Asel T. Jusupova3
1Department of Neurology and Neurosurgery, Kyrgyz State Medical Institute of Retraining and Advanced Training named after S. B. Daniyarov, Bishkek, Kyrgyz Republic
2Department of Special Clinical Disciplines, International School of Medicine of International University of Kyrgyzstan, Bishkek, Kyrgyz Republic
3Department of Neurology and Clinical Genetics, Kyrgyz State Medical Academy named after I. K. Akhunbaev, Bishkek, Kyrgyz Republic
Abstract
Objective: Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease affecting both central and peripheral motor neurons, presenting a significant challenge in modern neurology. This article discusses the multifactorial nature of ALS, its prevalence, and the absence of effective treatment. Clinical manifestations include damage to upper and lower motor neurons, with varied courses influenced by genetic mutations. Atypical forms, such as ALS with frontotemporal dementia, pose diagnostic challenges due to cognitive symptoms that may precede motor impairments. The study emphasizes the importance of neurophysiological and cognitive diagnostic methods in identifying associated conditions.
Case presentation: The article presents a clinical observation of a 51-year-old patient with a rare combination of ALS and frontotemporal dementia. The patient exhibited a progressive decline in motor function, cognitive impairments, and behavioral changes. Detailed clinical descriptions, including neurological and neuropsychological assessments, highlight the complexity of the case. Instrumental studies, such as magnetic resonance imaging and electomyography, revealed atrophy in frontotemporal areas and anterior horn involvement, corroborating the diagnosis. Treatment involved edaravone and B vitamins, resulting in a modest improvement in motor function.
Conclusion: The case underscores the rarity of combined ALS and frontotemporal degeneration, prompting further research and dynamic monitoring. While current interventions can slow disease progression, effective treatments for ALS remain elusive. The coexistence of frontotemporal dementia complicates the disease course, necessitating ongoing clinical attention and research efforts.
Key words: amyotrophic lateral sclerosis, frontotemporal dementia, atypical form, obesity treatment, eating disorder

Introduction
Amyotrophic lateral sclerosis (ALS) is a severe multifactorial neurodegenerative disease, and characterized by combined damage to central and peripheral motor neurons (1, 2). ALS is one of the pressing problems of modern neurology, which is associated with the defeat of mainly people of working age leading an active social life, the progressive course of the disease and the inevitability of a fatal outcome (3). The prevalence of ALS is 3–5 per 100 thousand of population, and the incidence is 1–2 per 100 thousand of population per year (4, 5).
Despite advancements in ALS research, the etiology and pathogenesis of the disease, as well as the factors causing uncontrolled motor neuron death, remain largely unexplained. Consequently, no preventive measures or effective treatments are available (4, 6, 7). Clinical manifestations of ALS include damage to both upper and lower motor neurons, varying in location, age of onset, rate of progression, and presence of extra-motor symptoms (1, 7). Different disease courses are observed depending on various patient mutations.
Considering the rare occurrence of sporadic cases of ALS in combination with frontotemporal dementia, we present our own clinical observation.
Case report
Patient A., a 51-year-old male with a body mass index (BMI) of 23.5, was hospitalized in December 2021 with complaints of weakness and limited movement in the arms, nasality, difficulty swallowing, choking while eating, muscle twitching, memory loss (especially for recent events), and weight loss. Relatives noted changes in his personality, a reduced range of interests, and speech difficulties.
Between 2018 and 2021, the patient lost 30 kg while on various diets for obesity. In March 2021, his relatives noticed memory decline for current events, repetitive speech, and cessation of reading books. By July 2021, he experienced weakness in the right arm, which later progressed to the left arm, along with involuntary muscle twitching, voice changes, and difficulty swallowing. By November 2021, he could not recognize familiar people, faced increased hand weakness, and had speech difficulties.
Diagnostic workup: After the initial examination and comprehensive testing, including neuroimaging and neuropsychological assessments, the patient, was diagnosed with ALS based on the international El Escorial criteria. Simultaneously, a diagnosis of frontotemporal dementia (FTD) was established using the diagnostic criteria for the behavioral variant of FTD, as developed by the international consensus of the working group in 2011. These criteria confirmed that the patient exhibited progressive changes in personality and behavior along with cognitive impairment, as evidenced by neuropsychological tests and characteristic changes on magnetic resonance imaging (MRI). Other conditions, such as Alzheimer's disease, vascular dementia, and other neurological and psychiatric disorders, were excluded.
General condition: The patient’s general condition is relatively satisfactory. The skin and visible mucous membranes are pale pink. Vesicular breathing is present. Breathing rate is 20 per minute, with no wheezing. Heart sounds are muffled but rhythmic. Blood pressure is 130/80 mmHg. Heart rate is 76 per minute. The abdomen is soft and painless. Stool and urine output are regular.
Neurological status: The patient is conscious, oriented in place and time, but exhibits reduced memory for current and distant events and a lack of awareness of his condition. Slow thinking and closed behavior are noted. Palpebral fissures are identical. Pupils are equal and reactive to light. Full movement of the eyeballs is present. The face is symmetrical. The tongue is midline with fibrillary twitching but no atrophy. Dysarthria, dysphagia, and dysphonia are present. Palatine and pharyngeal reflexes are absent. Tendon reflexes from the arms are uniformly reduced, while those from the legs are exaggerated with expanded reflexogenic zones. Muscle tone is reduced in the proximal upper extremities. Muscle strength is reduced in the arms: 1.0-1.5 in the proximal sections, 3 in the distal sections. Muscle strength in the legs is unchanged. Symmetrical muscle wasting is observed in the upper shoulder girdle and hands, with constant fasciculations. No sensory disorders are identified. Palmar-mental reflex is present bilaterally. No meningeal signs are observed. Coordination tests show an inability to perform the finger-nose test, but the knee-heel test is performed satisfactorily. Pelvic organ functions are not impaired.
Neuropsychological status: The patient, aged 51 with higher education, exhibits a negative attitude towards examination, with aggressive reactions. He is in apathy and requires care from his children. His son reports a significant decline in cognitive and executive functions over the past year. The patient shows signs of rhinorrhea and bent posture, looks older than his age, and has difficulty performing cognitive tests.
Laboratory and imaging findings: General and biochemical blood tests are within normal range. Chest X-ray and electrocardiogram are unremarkable. Computed tomography (CT) of the cervical spine (October 29, 2021) shows degenerative-dystrophic changes with hernial protrusions at the levels of C3-4, C5-6, and C6-7. MRI of the brain (October 22, 2021) indicates encephalopathy with atrophic changes in the frontotemporal lobes and hippocampus, with gliosis and mixed hydrocephalus (Fig. 1). Electromyography (EMG) of the upper and lower extremities (December 29, 2021) shows increased motor unit potential duration and spontaneous activity, suggesting anterior horn involvement.
The patient was treated with edaravone 60 mg intravenously once a day for 14 days (in the future it is recommended to take 2 more courses with a break of 14 days), B vitamins 2.0 IM. On the 8th day after the start of treatment, the patient showed positive dynamics in the form of an increase in the range of active movements in the proximal parts of the upper extremities.
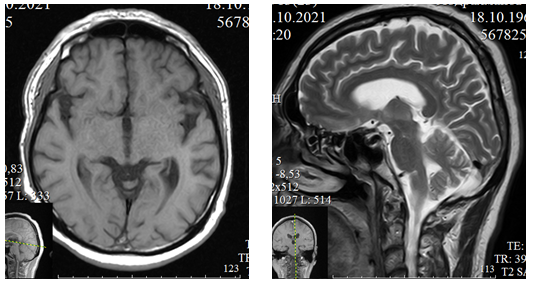
Figure 1. MRI of the brain of patient A., 51 years old, in T1 (axial section) and T2 (sagittal section) modes reveals atrophic changes in the frontotemporal areas
Discussion
The clinical case presented above aroused particular interest among clinicians due to the rare occurrence in clinical practice of two neurodegenerative nosologies - ALS and FTD. The patient's clinical manifestations are represented by progressive symptoms of damage to both central and peripheral motor neurons in combination with cognitive impairment. The disease began for no apparent reason; at the onset of the disease, cognitive impairments, predominantly of amnestic nature, prevailed, followed by the addition of emotional and affective disorders, symptoms of damage to the pyramidal system and bulbar symptoms. Eating disorders in this patient are also noteworthy. Such manifestations are associated with the involvement of the right medio- and orbitofrontal parts of the brain in the pathological process (2, 10).
The results of instrumental studies - MRI of the brain and EMG of the limbs, are represented by atrophy of the frontotemporal cortex of the brain and involvement of the anterior horns in the process, which confirm the diagnosis established for the patient. During the treatment, the patient showed positive dynamics in the form of a slight increase in muscle strength in the proximal parts of the upper extremities.
Conclusion
Thus, the manifestations of ALS in the patient were characterized by the development of bulbar disorders in the form of dysarthria, dysphonia and dysphagia, pyramidal disorders - severe upper paraparesis with predominant involvement of the proximal parts, muscle wasting, hyporeflexia and fascicular twitching in them, with characteristic changes on EMG. Symptoms of frontotemporal degeneration are represented by developed progressive cognitive and behavioral disorders, which are caused by the involvement of the frontotemporal cortex in the pathological process, confirmed by atrophic changes in these areas on MRI of the brain. ALS and frontotemporal dementia may co-occur, highlighting the importance of comprehensive assessments in patients with overlapping symptoms. Early and accurate diagnosis of both conditions is critical for appropriate management and care planning. Awareness of the continuum of frontotemporal dementia and motor neuron diseases can aid in better understanding and diagnosing such cases. Regardless of the timeliness of the diagnosis, unfortunately, today there is no effective treatment for ALS. Modern drugs can only slow down the rate of progression of the disease, thereby extending the patient’s life and improving its quality. The combination of two neurodegenerative diseases aggravates the course of the disease and requires further dynamic monitoring of the patient.
Ethics: Informed consent for all procedures and participation in this case study was obtained from the patient. The patient and their family were provided with comprehensive information regarding the nature of the study, the procedures involved, and the potential risks and benefits. All ethical guidelines and regulations were followed to ensure the patient's rights and well-being were protected throughout the study.
Peer-review: External and internal
Conflict of interest: None to declare
Authorship: N.T.D., E.B.B., B. B. K., I. A.S., and A.T. J. equally contributed to case management and preparation of manuscript for publication. All authors fulfilled authorship criteria.
Acknowledgements and funding: None to declare
Statement on A.I.-assisted technologies use: Authors declared they did not use AI-assisted technologies in preparation of this manuscript
References
| 1.Koberskaya NN, Grishina DA, Yakhno NN. Amyotrophic lateral sclerosis syndrome - Alzheimer's dementia. Russ Neurol J 2021; 26: 17-24. https://doi.org/10.30629/2658-7947-2021-26-2-17-24 |
||||
| 2.Kovrazhkina EA, Razinskaya OD, Gubsky LV. Clinical polymorphism of amyotrophic lateral sclerosis. J Neurol Psych 2017; 8: 4-10. https://doi.org/10.17116/jnevro2017117814-10 PMid:28884711 |
||||
| 3.Levitsky GN, Levitsky AS, Gilod VM. Mental disorders in patients with amyotrophic lateral sclerosis and members of their families. J Neurol Psych S.S. Korsakov 2015; 115: 64-7. https://doi.org/10.17116/jnevro20151152164-67 PMid:26081327 |
||||
| 4.Egorkina OV, Gaponov IK. Clinical approach to the treatment of neurodegenerative diseases with dementia. Intern Neurol J 2007; 1: 111-7. | ||||
| 5.Armstrong RA. euronal cytoplasmic inclusions in tau, TDP-43, and FUS molecular subtypes of frontotemporal lobar degeneration share similar spatial patterns. Folia Neuropathol 2017; 55: 185-92. https://doi.org/10.5114/fn.2017.70482 PMid:28984110 |
||||
| 6.Burrell JR, Halliday GM, Kril JJ. The frontotemporal dementia-motor neuron disease continuum. Lancet. 2016;388(10047):919-931. https://doi.org/10.1016/S0140-6736(16)00737-6 PMid:26987909 |
||||
| 7.Strong MJ, Abrahams S, Goldstein LH. Amyotrophic lateral sclerosis-frontotemporal spectrum disorder (ALS-FTSD): revised diagnostic criteria. Amyotroph Lateral Scleral Frontotemporal Degener. 2017;18(3-4):153-174.1, 9, 12 https://doi.org/10.1080/21678421.2016.1267768 PMid:28054827 PMCid:PMC7409990 |
||||
| 8.Bannwarth, S., Ait-El-Mkadem, S., Chaussenot, A., Genin, E.C., Lacas-Gervais, S., Fragaki, K., et al. (2014). A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. 9.Beeldman E, Govaarts R, de Visser M. Progression of cognitive and behavioral impairment in early amyotrophic lateral sclerosis. J Neurol Neurosurg Psych 2020; 91: 779-80. | ||||
| 10.Yakhno NN, Golovkova MS, Preobrazhenskaya IS, Zakharov VV. ALS-dementia syndrome of frontal type. Neurol J 2002; 7: 12-7. | ||||
| 11.Rascovsky K, Hodges JR, Knopman D. Sensitivity of revised diagnostic criteria for the behavioral variant of frontotemporal dementia. Brain 2011; 134: 2456-77. https://doi.org/10.1093/brain/awr179 PMid:21810890 PMCid:PMC3170532 |
||||
| 12.Castelnovo V, Canu E, Riva N. Progression of cognitive and behavioral disturbances in motor neuron diseases assessed using standard and computer-based batteries. Amyotroph Lateral Scleral Frontotemporal Degener 2021; 22: 223-36. https://doi.org/10.1080/21678421.2020.1867179 PMid:33463386 |
||||
Copyright

This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.
AUTHOR'S CORNER
