Mechanistic relationship between obesity-induced inflammation triggering endothelial dysfunction and the initiation of atherosclerosis development
REVIEW
Mechanistic relationship between obesity-induced inflammation triggering endothelial dysfunction and the initiation of atherosclerosis development
Article Summary
- DOI: 10.24969/hvt.2024.542
- CARDIOVASCULAR DISEASES
- Published: 19/01/2025
- Received: 12/11/2024
- Revised: 20/12/2024
- Accepted: 21/12/2024
- Views: 13140
- Downloads: 3881
- Keywords: Obesity, inflammation, reactive oxygen species, adipose tissue, endothelial dysfunction, atherosclerosis
Address for Correspondence: Elhadi H Aburawi, Department of Pediatrics, College of Medicine and Health Sciences, United Arab Emirates University, Al-Ain, Abu Dhabi, P.O. Box 15551, United Arab Emirates
E-mail: e.aburawi@uaeu.ac.ae Phone: +971 7137462 Fax: +971 3 7672022
ORCID: Elhadi H Aburawi - 0000-0001-5200-9048
Richard L. Jayaraj1,2, Elhadi H Aburawi1*
1Department of Pediatrics, College of Medicine and Health Sciences, UAE University, Al-Ain P.O. Box 15551, United Arab Emirates
2Institute of Sciences in Emergency Medicine, Department of Emergency Medicine, Guangdong Provincial People's Hospital (Guangdong Academy of Medical Sciences), Southern Medical University, Guangzhou, China
Abstract
Obesity is a multifactorial, chronic inflammatory illness that affects individuals of all ages; it is linked to several cardiovascular and metabolic syndromes. Excessive accumulation of fat that impairs metabolic processes is a key feature of obesity. Globally, more than 1.9 billion adults are overweight or obese, and the number of people affected by obesity is increasing. Obesity contributes to cardiovascular disease (CVD) and metabolic disease (MD) through processes such as inflammation, hyperglycemia, and dyslipidemia, as well as metabolic syndrome and obstructive sleep apnea. Furthermore, alterations in adipose tissue composition in obesity play a major role in the production of inflammatory cytokines that promote endothelial dysfunction (ED). Endothelial dysfunction is the first step in the mechanisms underlying obesity-related complications, such as atherosclerosis, which is a major complication of obesity. A growing body of evidence suggests that obesity-related ED makes a significant contribution to the development of CVD and MD.
Several mechanisms explain the association between obesity and atherosclerosis. The aim of this review is to summarize the effects of obesity-related inflammation on ED and its progression to atherosclerosis, focusing on cellular senescence, vascular aging, epigenetic modifications, reactive oxygen species, vascular calcification, and gut microbiota. In addition, we are also exploring new therapeutic strategies to reverse ED to prevent CVD and MD. Therefore, understanding the mechanisms underlying obesity-induced ED and its effects on atherosclerosis is crucial for developing therapeutic interventions.
Key words: Obesity, inflammation, reactive oxygen species, adipose tissue, endothelial dysfunction, atherosclerosis.
Introduction
Obesity is a major public health issue worldwide. Excessive body fat storage without energy conversion plays a major role in obesity. Healthcare expenses, morbidity, and mortality are all substantially affected by obesity. It has reached epidemic proportions in many countries, affecting not only adults but also children and adolescents (1).
Cardiovascular (CV) disease (CVD) is the leading cause of death worldwide. Obesity is the most important risk factor for the development of coronary artery disease (CAD) (2). Some high-income nations have already experienced an initial obesity pandemic wave, whereas some low- and middle-income nations have experienced a second wave. More than 25% of adults in some middle-income countries, such as South Africa, Libya, Egypt, Mexico, and Argentina, are reportedly afflicted with obesity. Compared to the rate of obesity prevalence just 20 years ago, this is a significant increase (3).
Obesity increases the risks of atherosclerosis (AS), stroke, type 2 diabetes mellitus (T2DM), and cardiovascular events (Fig. 1) (4, 5). According to the Global Burden of Disease survey reported in 2020, 4 million deaths, of which 2.7 million were CV, were attributable to high body mass index (BMI). (6). Obesity has attracted considerable attention as a global public health concern.
Graphical abstract

One type of obesity, known as metabolically healthy obesity (MHO), lacks cardiometabolic risk factors, such as hypertension, T2DM, insulin resistance and dyslipidemia (7). Individuals with the MHO phenotype do not seem to develop AS (8).
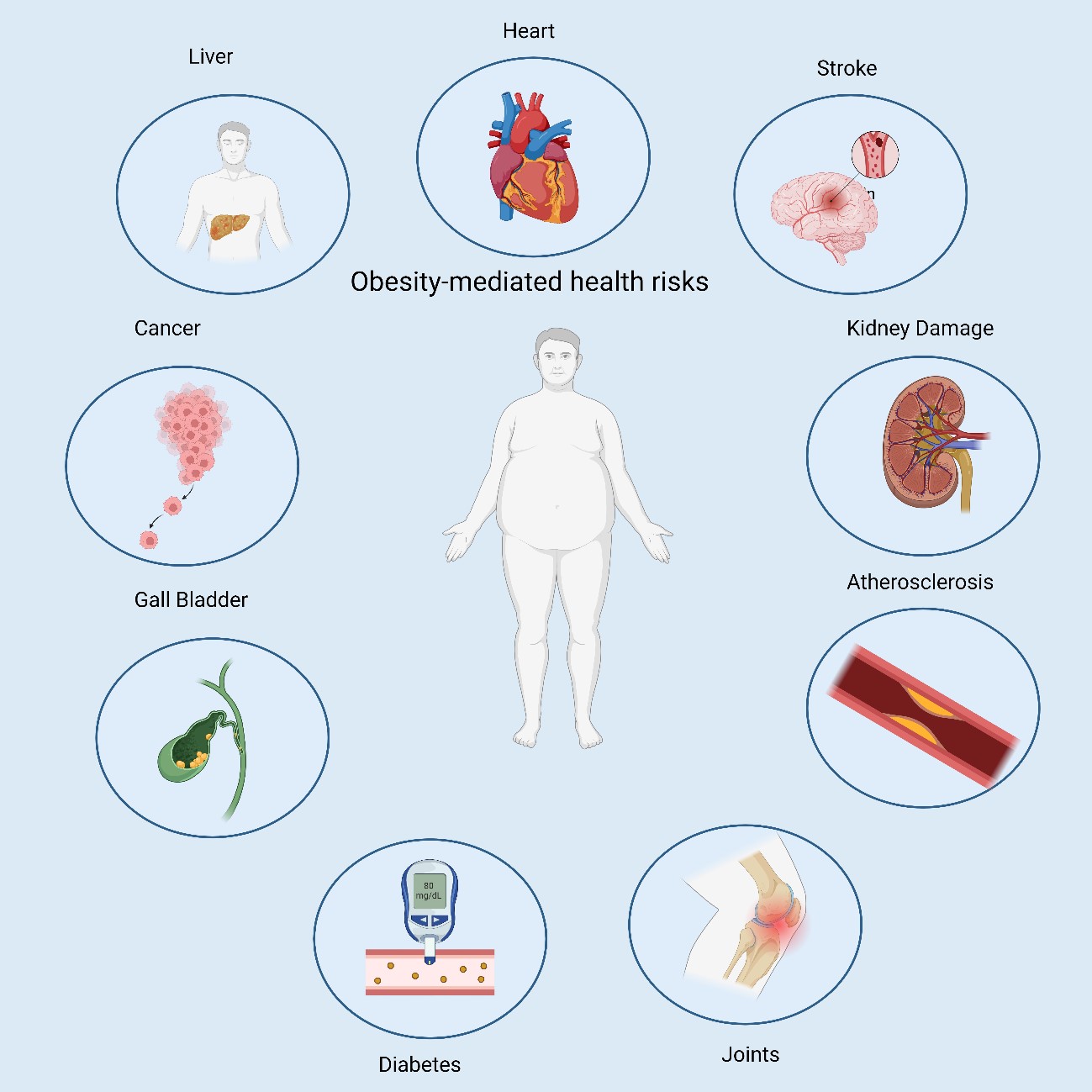
Figure 1. Comorbidities associated with obesity
Although various factors such as oxidative stress, loss of autophagic flux, macrophage NOD-like receptor protein 3 (NLRP3), inflammasome activation, adipokine imbalance, altered gut microbiome, and vascular inflammation are involved in obesity-induced AS, endothelial dysfunction (ED) plays a major role in CVD progression. In addition to genetic and environmental factors, obesity has been suggested to play a major role in the development of ED and AS (9).
Physiologically, vasoconstricting and vasodilating factors are secreted and released from ECs in a balanced manner (Fig. 2). Obesity-mediated AS disrupts this balance, thereby promoting the development of vascular ED and organ damage (10). Atherosclerosis commences in childhood when cholesterol esters from macrophage foam cells are ingested, and the deposition of these esters on the wall surfaces of blood vessels leads to arterial wall thickening. Eventually, fatty streaks are formed owing to further lipid accumulation, which appears in young adults (11, 12).
Recently, much attention has been paid to the mechanisms underlying obesity-mediated ED, which have substantial implications for the prevention and treatment of CVD. The current review focuses on elucidating the pathophysiological mechanisms by which obesity-induced ED leads to AS and identifying therapeutic targets for the treatment of obesity.
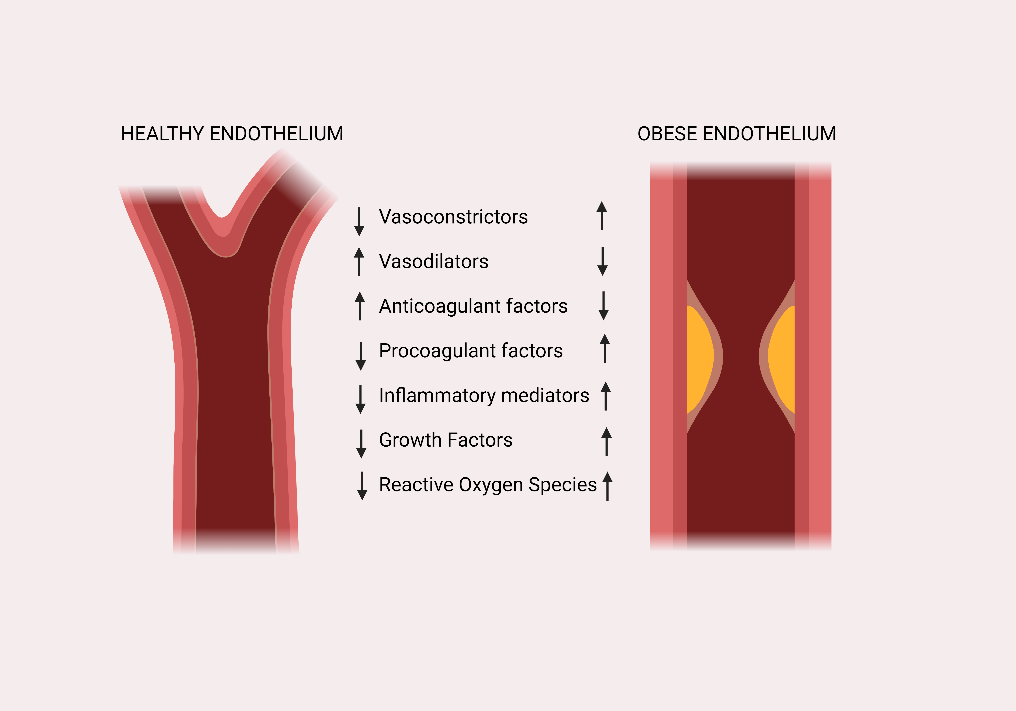
Figure 2. Differences in function of endothelial cells in healthy patients and patients with obesity
An overview of the mechanisms and their importance
Signaling mechanisms that contribute to ED in obesity-induced AS include autocrine, paracrine, and endocrine signaling. Studies have shown that tumor necrosis factor alpha (TNF-α) induces reactive oxygen species (ROS) production in small visceral fat arteries from patients with obesity, reducing nitric oxide (NO) availability (13).
Using in vivo and ex vivo myographic techniques, a reduction in endothelial function in the vascular region has been demonstrated, and the potential mechanisms include ROS-mediated rather than endothelial nitric oxide synthase (eNOS).-mediated dysfunction (13).
Classically, proinflammatory cytokines released by adipocytes are believed to play a major role in ED. Major adipokines generated by fat cells, such as resistin, leptin, and adiponectin, play important roles in inflammation. Leptin, one of the crucial proteins produced by adipocytes, enhances the secretion of interleukin-6 (IL-6); furthermore, TNF-α directly or indirectly increases the levels of ROS, resulting in ED (14). Additionally, obesity damages the endothelial glycocalyx, which regulates NO production by responding to blood vessel forces. It was found that by thinning of the glycocalyx in mice and humans because of obesity, flow-mediated vasodilation was prevented in mesenteric arteries (15). Hindrance in endothelial properties causes the vascular endothelium to adopt a proinflammatory, prothrombotic, and proatherogenic phenotype, resulting in the pro-oxidation of mitogens, leukocyte adhesion, and platelet activation, as well as an increase in the levels of vasoconstriction factors, decreased synthesis of endothelium-derived hyperpolarizing factors (EDHFs), and impaired endothelial NO production (16) (Fig. 3).
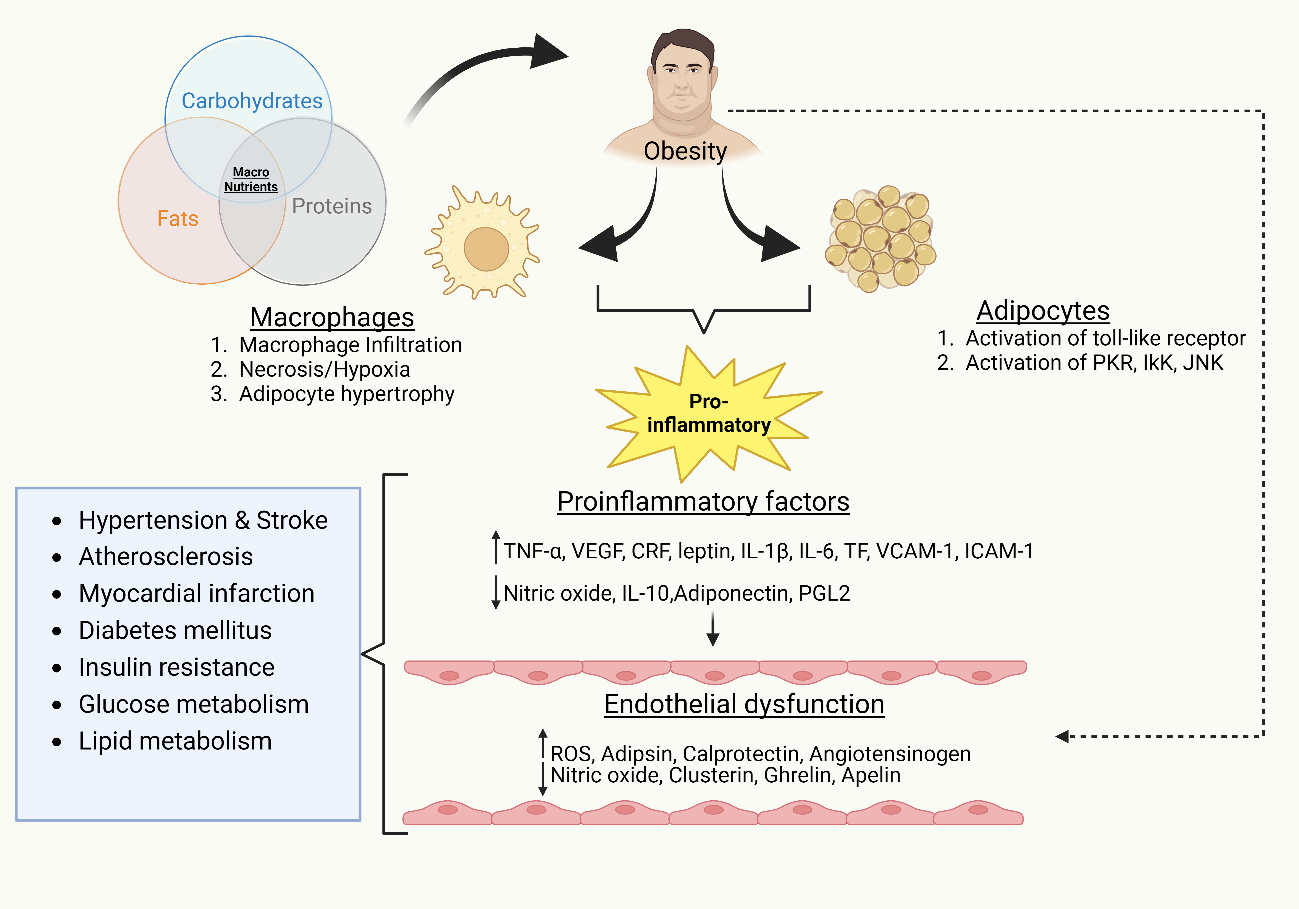
Figure 3. Pathological molecular mechanisms of obesity-induced endothelial dysfunction contributing to other diseases
Endothelial dysfunction causes CVD progression in obesity by activating adhesion molecules, leukocyte proliferation, and transmigration (17). Furthermore, dysfunctional adipocytes secrete angiotensinogen from the renin–angiotensin system (RAS), which causes its overexpression in RAS, enhanced ROS production, and atherogenic and thromboembolic potential of ECs (18). Adipose tissue is typically composed primarily of adipocytes and pre-adipocytes, with only minimal leukocytes. However, during obesity, the configuration and function of adipose tissue are largely altered. Endothelial cells secrete endocan (also known as endothelial cell-specific molecule-1), a soluble proteoglycan. The expression of endocan can be upregulated by inflammatory factors like TNF-α and IL-1β, which can then influence the expression of cell-adhesion molecules like vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1). These molecules are crucial for promoting leukocyte migration and the inflammatory response. Elevated plasma endocan levels are considered a possible inflammatory marker associated with CVD, which reflect endothelial activation and dysfunction (19).
Furthermore, endoglin, also known as CD105 or transforming growth factor beta receptor-3 (TGF-β receptor III), a homodimer transmembrane glycoprotein, is one of the ED inducers. Under several circumstances involving endothelial damage, activation, inflammation, and endothelial senescence, soluble endoglins enter the systemic circulation. Furthermore, their levels increase during the early stages of AS and decreased in the later stages (20). Consequently, endoglin and endocan have been studied as potential biomarkers of ED and AS.
Adipose tissue alteration during obesity
White and brown adipose tissue are typical of mammals. Most of the mammalian adipose tissue, which is labelled as the area of energy storage, is white (21).. White adipose tissue acts as a layer of thermal insulation, energy reservoir, mechanical protection tissue, and endocrine organs. Brown adipose tissue serves as an energy combustion site that maintains a stable body temperature. White adipose tissue accounts for 30–40% of the total body weight in lean women and 15–25% in lean men, whereas brown adipose tissue accounts for only 1–2% of fat in adult humans (22).. In newborns, brown adipose tissue is typically elevated and works well in controlling energy expenditure and body temperature during non-shivering thermogenesis induced by uncoupling protein-1 (UCP1). (23). Visceral adipose tissue secretes more adipokines than subcutaneous adipose tissue, and these adipokines modulate adipocyte differentiation, lipid and glucose metabolism, neuroendocrine and cardiovascular functions, inflammatory processes and immunity (24).
Adipokines, chemokines, and cytokines, which are primarily elevated in obesity, are among the inflammatory factors produced by the adipose tissue, and an imbalance between pro- and anti-inflammatory factors plays a major role in obesity-mediated AS (25) (Fig. 4).
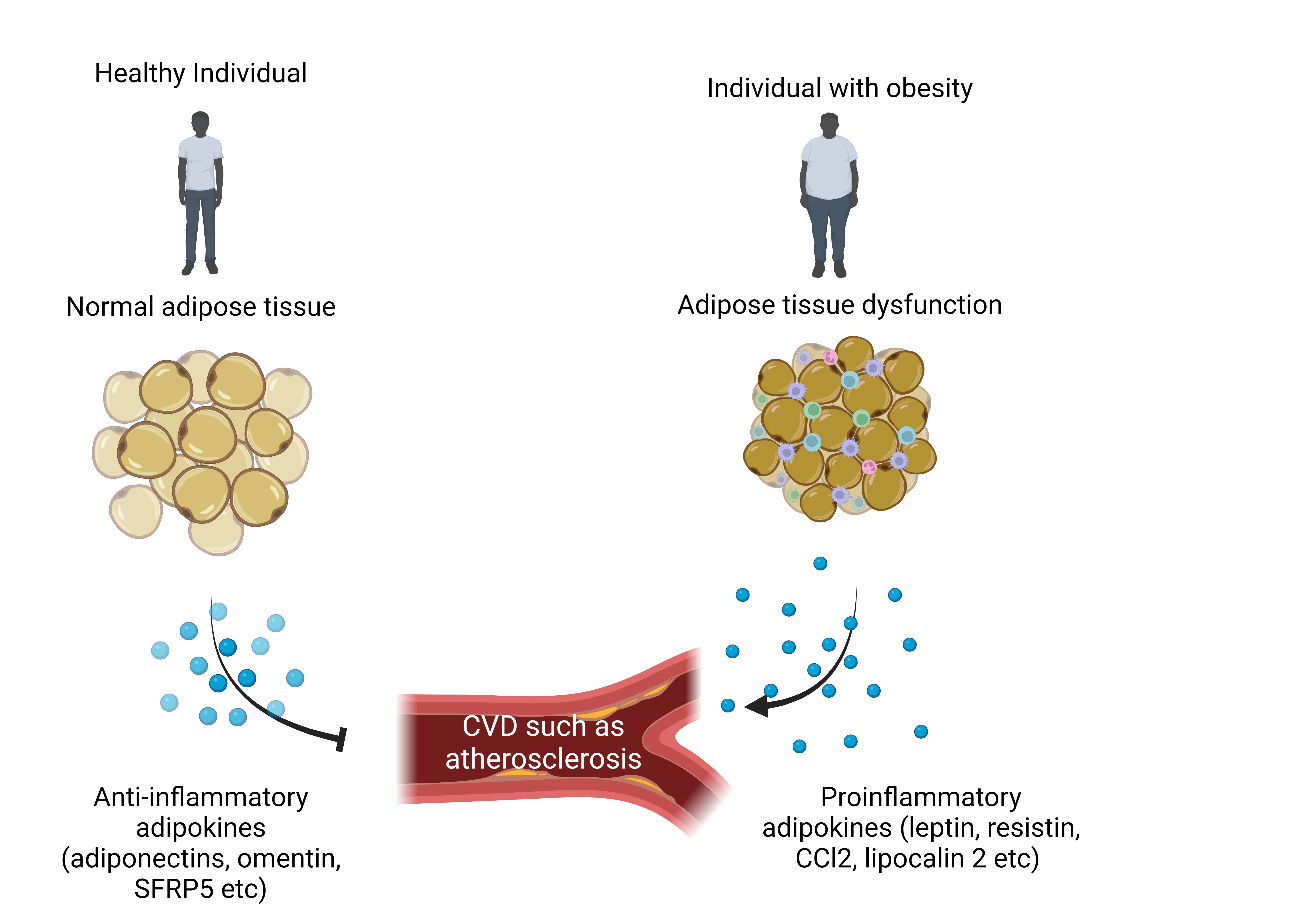
Figure 4. Physiological and pathological role of adipose tissue contributing to atherosclerosis
In addition, a different subset of UCP1-positive cells, called beige adipocytes, is prevalent in white adipocytes. Under the activation of sympathetic adrenergic receptors or cold stimulation, white adipocytes differentiate into brown-like adipocytes through a phenomenon called browning. Experimental reports have demonstrated the antiobesity and antidiabetic properties of beige adipocytes; consequently, they have garnered much attention for their therapeutic potential (26).
Adipose tissue inflammation is typically characterized by the infiltration of immune cells, including B and T lymphocytes, neutrophils, and macrophages. The primary cause of inflammation linked to ED is macrophage infiltration of adipose tissues.
Macrophage-specific genes, such as monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein-1 (MIP-1), F4/80, CD11b, and CD68, have been found to be enhanced in the obese mice adipose tissues.
Adipocyte size, F4/80 expression, and body mass were significantly correlated in adipose tissues after immunohistopathological analysis (27).
The composition, size, and structure of adipose tissue play a major role in the pathogenesis of obesity-induced CVD, such as AS. Adiposopathy or sick fat, a recently coined term for qualitative fat abnormalities, varies depending on an individual’s lifestyle and physical activity (28). Clinical studies using cross-sectional abdominal imaging have shown that visceral adiposity has a stronger relationship with the incidence of systemic cardiometabolic rather than with subcutaneous fat quality (29). Although visceral fat contributes to only 5–20% of the total body fat mass compared with subcutaneous fat (≈ 80%)., it contains an abundant source of IL-6, TNF-α, c-reactive protein in patients with abdominal obesity than in those with peripheral obesity (30). In addition, MRI or CT studies of the fat compartment demonstrated that systemic ED is significantly associated with visceral fat volume compared to subcutaneous fat volume, and gene-expression studies of visceral fat showed enhanced expression of antiangiogenic, proinflammatory, and oxidative stress-related genes (31-33).
Endothelial structure and function
The EC monolayer that lines the lumen of all blood vessels is known as endothelium. These cells serve as biocompatible barriers between all tissues and the flowing blood, which are both protective and insulating. The layers are arranged as follows: tunica intima, made up of ECs; the tunica media, which forms vascular smooth muscle cells (VSMCs); and tunica adventitia, which is an elastic lamina comprising terminal nerve fibers around connective tissues (Fig. 5). The endothelial glycocalyx is the apical surface of the endothelium and is composed of glycoproteins, proteoglycans, and glycosaminoglycan chains (34, 35). Together, endothelial glycocalyx, secreted proteoglycans, and adsorbed plasma proteins form the endothelial surface layer. The ECs play a crucial role in homeostasis and arterial smooth muscle relaxation, and, consequently, vascular tone maintenance (36).
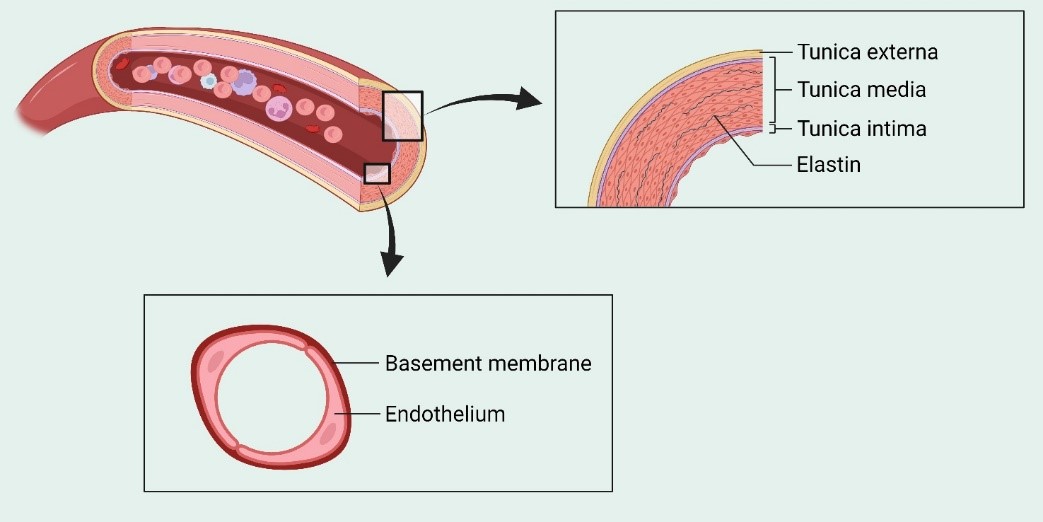
Figure 5. Structure of endothelium
The strategic location of endothelium helps to ‘sense’ changes in hemodynamic forces and blood-borne signals and ‘respond’ by releasing various autocrine and paracrine substances. Under normal physiological conditions, the balanced release of these bioactive compounds helps in maintaining vascular homeostasis (Fig. 6). Endothelium plays a major role in the modulation of thrombogenicity, angiogenesis, vascular tone, inflammation, and dynamic permeability through receptor-mediated mechanisms and the balance between the release of autacoids, such as prostacyclin, endothelin-1, and angiotensin II (37-39).
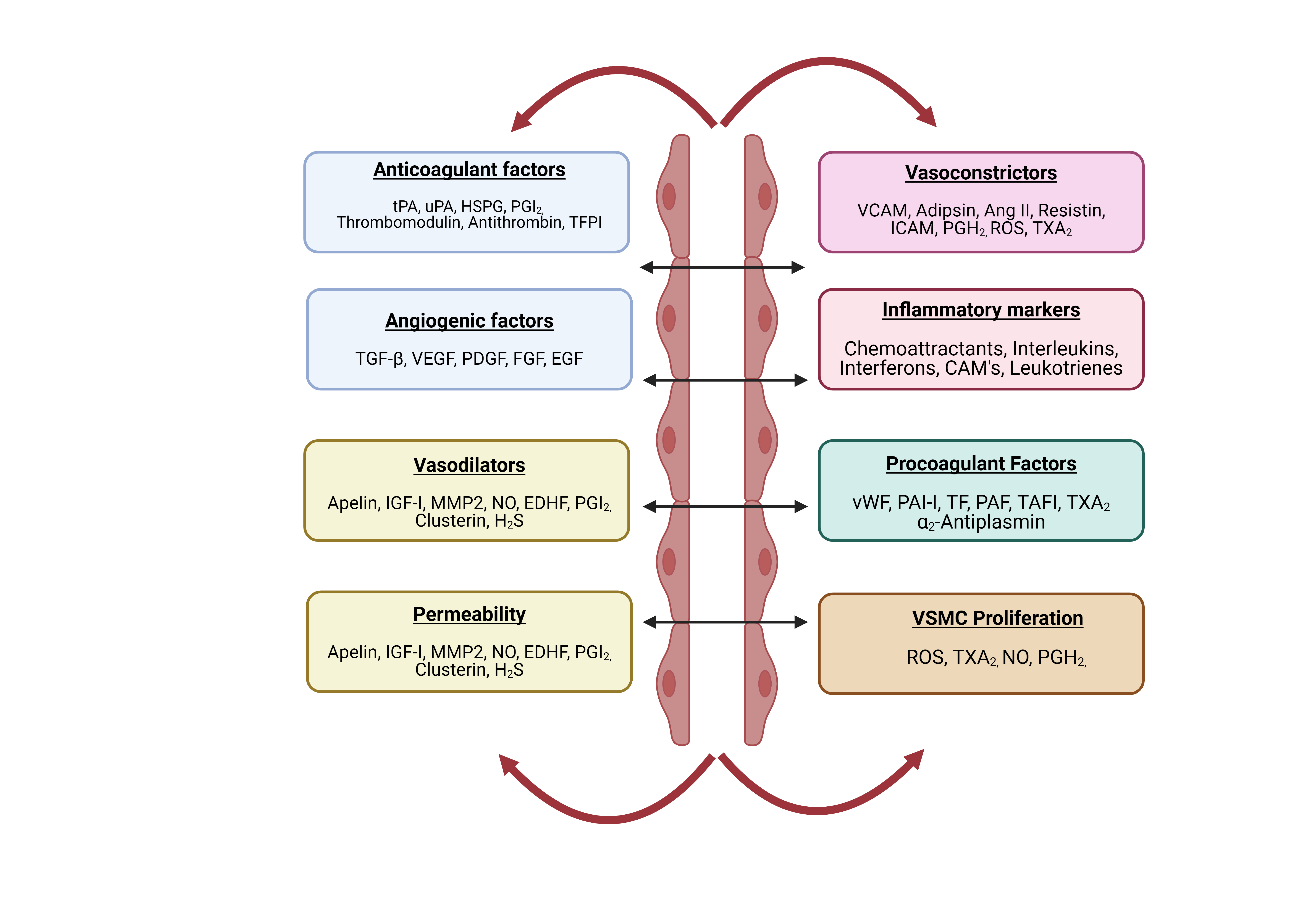
Figure 6. Important function of endothelial cells in regulating vascular homeostasis
The vascular endothelium produces prostacyclin (PGI2) and NO upon exposure to ecto-adenosine diphosphate (ADP)-ase, CD73, and CD39, and prostaglandin E2 is promoted by glycosaminoglycans on the surface of ECs, thereby preventing platelet adhesion and aggregation. The endothelium also interacts with antithrombin III (ATH III), tissue factor pathway inhibitors (TFPIs), thrombomodulin (TM), and tissue factor (TF) to control adhesion and coagulation factor activity. In addition, the endothelium regulates urokinase plasminogen activator-1 (uPA-1), tissue-type plasminogen activator-1 (tPA-1), and inhibitors of uPA-1 and PAI-1. These mechanisms are influenced by the secretion of regulatory mediators, such as NO, endothelin-1 (ET-1), t-PA, PAI-1, prostanoids, adhesion molecules, angiotensin II (Ang II), von Willebrand factor (vWF), and cytokines. Endothelial cells secrete bioactive substances that control and maintain the structures and functions of intact blood vessels by balancing oxidative and antioxidative agents, VSMC proliferative and antiproliferative agents, inflammatory and anti-inflammatory factors, the blood coagulation cascade, vascular dilations and contractions, and the fibrinolytic system (40).
The endothelium exhibits anticoagulant activity via the synthesis of heparin sulfate proteoglycan, TM, and secretion of TFPIs. Further binding of TM to thrombin stimulates protein C, which inhibits factor VIII and factor V blood clotting. In addition, heparin-like molecules act as cofactors for ATH III, an anticoagulant molecule, whereas the complex activities of Factor Xa and TF-Factor VIIa are hindered by TFPIs.
Endothelial tissue plasminogen activator initiates fibrinolytic mechanisms, whereas NO and prostacyclin (PG12) are produced to control endothelial antiplatelet activities (41). The calcium–calmodulin complex activates eNOS to produce NO, L-arginine, nicotinamide adenine dinucleotide phosphate (NADPH), and tetrahydrobiopterin (BH4) as substrates. Furthermore, suppression of vasoconstriction and initiation of vasodilation are achieved by the absorption of NO by VSMCs, which then form a complex with the hetero-iron group of guanylate cyclase. This complex, in turn, forms cyclic guanosine monophosphate (cGMP), activates cGMP-dependent protein kinase, and elevates Ca2+ levels in the cytosol within VSMCs.
Nitric oxide is a potent endothelium-derived vasodilator that possesses antiproliferative, antiplatelet, anti-inflammatory, and permeability-reducing properties. In addition, NO reacts with Kruppel-like factor 2 and EDHFs to enhance arterial vasodilation. Conversely, NO also inhibits MCP-1 and VCAM-1, thereby downregulating nuclear factor kappa B (NF-κB) expression (21).
Influence of endothelial dysfunction on cellular senescence and vascular aging
Atherosclerosis is an important cause of death in the Western world, which is associated with smoking, dyslipidemia, and hypertension as risk factors (48, 49). In addition to disease-related ED, age-related ED plays an important role in CVD owing to its proinflammatory, vasoconstrictive, and prothrombotic environment(42), thereby disrupting vascular homeostasis.
Senescent cells are distinct in that they eventually cease to proliferate but do not eventually die, as they occur in a normal physiological process. Instead, the cells release substances that cause inflammation. Endothelial senescence is triggered by different senescence stressors, such as DNA damage (43), oxidative stress (44), oncogenic activation, and mitochondrial dysfunction (45).
Aged ECs show increased endothelin-1 (ET-1) expression, inflammation, and apoptosis, with attenuated endothelial NO production.
This results in the impairment of vessel tone, vascular integrity, and angiogenesis, resulting in the progression of CVD (46).
Adenosine monophosphate-activated kinase (AMPK), a mammalian target of rapamycin (mTOR), is a critical sensor of cellular homeostasis that influences endothelial senescence (47). A few clinical studies have shown that age-mediated arterial stiffness increases systolic pressure and decreases diastolic pressure, resulting in pulse pressure elevation and aortic pulse wave velocity (48).
Altered Ca2+ signaling in EC plays a major role in ED. Enhanced Ca2+ signaling promotes endothelium-dependent vasodilation via prostacyclin, NO, and EDHF. Physiologically, ECs play a major role in increasing Ca2+ levels and hyperpolarization by activating small-and intermediate-conductance Ca2+-activated potassium channels that initiate electrical conductance between vascular smooth muscle cells and ECs (46). However, all these pathways are impaired during aging, resulting in vascular dysfunction (Fig. 7).
Peroxynitrite, hydrogen peroxide, and hydroxyl radicals are the sources of ROS that contribute to age-related ED. In addition to ROS production through peroxisomal β-oxidation of fatty acid, mitochondrial respiration, lipoxygenase and xanthine oxidase, ROS levels are also increased via the uncoupling of eNOS, which results in the formation of peroxynitrite (49). Isoforms of nicotinamide adenine dinucleotide phosphate oxidase (NOX), such as NOX1, NOX2, NOX4, and NOX5, contribute to ED, apoptosis, and inflammation in AS (50). In addition, oxidative stress generates oxidized phospholipids (OxPLs) from polyunsaturated diacyl and alkaline (en) ylacyl glycerophospholipids.
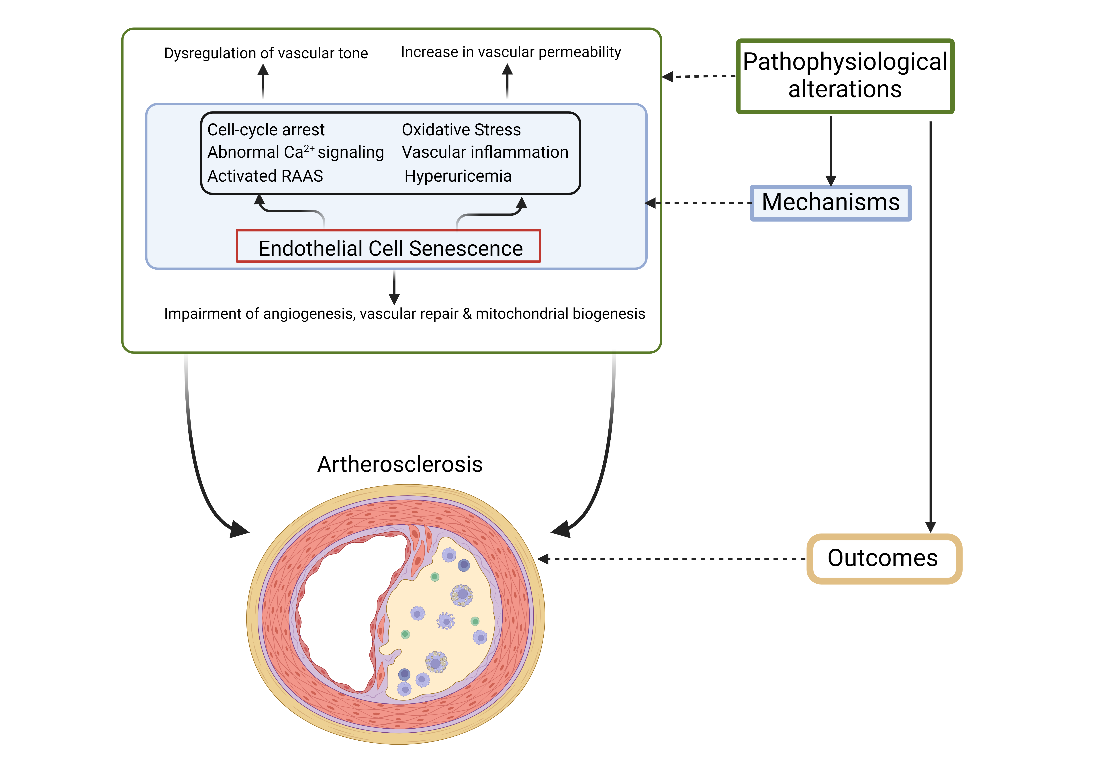
Figure 7. Pathophysiological mechanism of endothelial senescence resulting in atherosclerosis
Therefore, oxidative stress-induced cellular stress plays a major role in AS development. Various in vitro and in vivo studies have shown that OxPLs contribute to the development of AS. Briley et al. (51) demonstrated the presence of OxPLs in the atherosclerotic vessels of human lesions and in a hypercholesterolemia animal model. OxPLs contribute to the expression of proinflammatory cytokines in vascular ECs, thereby directly acting on leukocytes and influencing monocyte adhesion (52). Given their pertinent role in the development of numerous pathological conditions, OxPLs may function as biomarkers for the early detection of several CVDs, including AS.
Aging results in the enhanced expression of inflammatory cytokines, chemokines, and adhesion molecules in ECs. Interleukin-6, MCP-1, cellular adhesion molecules, IL-1β and TNF-α are key cytokines that are overexpressed because of initiation of NF-κB signaling resulting in endothelial dysfunction (50). In addition, activation of NLRP3 initiates oligomerization resulting in attracting procaspase-1, leading to production of ROS, chemokines and activation of NF-κB (53). Inhibition of NF-κB activation during prolonged TNF-α exposure prevents the premature senescence of ECs, thereby demonstrating a pathological association between inflammation and EC senescence (53, 54).
Oxidative stress and inflammation play major roles in the activation of the vascular renin–angiotensin–aldosterone system (RAAS). Atherogenesis is a critical process in atherosclerotic plaque formation and is influenced by the activation of angiotensin II, which further decreases NO generation. Inflammation, altered lipid metabolism, and endothelial injury can lead to atherogenesis. Of these, endothelial injury is the initial trigger leading to the infiltration and accumulation of multiple modified low-density lipoprotein (LDL) particles in the endothelium (55). The accumulation of modified LDL initiates the production of VCAM-1 and P&E-selectins, resulting in the recruitment of leukocytes to the endothelial space, thereby attracting inflammatory cells, which are influenced by MCP-1.
Furthermore, macrophages express scavenger receptor class A (SRA), cluster of differentiation 36 (CD36), which act through modified LDL internalization, resulting in inflammation and ROS (56).
The senescence of ECs leads to CVD, and uric acid levels are higher in aged women than in men (57). Aortic stiffness is closely associated with the inflammatory response and maladaptive immunity. Inhibition of aortic xanthine oxidase activity (increased because of the western diet) by allopurinol decreases aortic stiffness and impairs endothelial-dependent arterial relaxation (58). A large multicenter study by Neogi et al. demonstrated an association between serum uric acid levels and carotid plaques (59). They also found this association in men, even in the absence of other CVD risk factors and coronary calcifications.
Epigenetic modifications
Histone acetylation and DNA methylation are examples of epigenetic modifications that are post-replication chromatin alterations that do not affect basal nucleotide code (60). These processes are linked to several age-related diseases, such as AS, and are crucial for regulating gene activation and silencing (61). Therefore, epigenetic changes have been used as biomarkers of cellular senescence in vascular ECs. Prior research has identified specific changes in methyltransferase expression, suggesting that these changes are associated with hypomethylation of hypermethylated genomic regions. These genes are involved in lipid metabolism, proliferation, and apoptosis. Atherogenic lipoproteins promote DNA methylation and histone deacetylation (60). These results suggest a possible association between AS and epigenetic alterations. Histone acetylation and deacetylation play major roles in the development of AS with age, and these changes can affect VSMC proliferation, extracellular matrix (ECM) composition, and inflammation. Moreover, additional research has demonstrated that the mitochondrial gene p66Shc, which is recognized as a gene associated with longevity, stimulates histone acetylation and hypermethylation, causing an age-related increase in p66Shc synthesis and its consequent activation (62).
The Genetics of Healthy Aging Study demonstrated that genes encoding apolipoprotein E, forkhead box protein O1, IL-6, and Sirt6 are functionally associated with EC senescence (63). Among these, transcriptional expression and senescence were significantly correlated with argininosuccinate synthase 1, NOX4, aquaporin 1, p15, and p16. Telomere changes are a key mechanism in the senescence of vascular cells. Leukocyte telomere length is substantially correlated with vascular cell senescence, hypertension, and atherothrombotic events in a clinical study involving a cohort of 193 patients (≥70 years of age) (64). Therefore, telomerase activity controls both CVD and EC functions. Single-stranded RNAs of approximately 25 nucleotides, known as microRNAs (miRNAs), regulate the pathobiology of CVD and EC senescence. Overexpression of ASncmtRNA-2 induces replicative senescence in ECs and cell-cycle arrest in the G2/M phase, indicating a potential role for these non-coding RNAs in the regulation of EC senescence, vascular function, and CVD (65).
Impact of oxidative stress
Oxidative stress markers are highly correlated with fat accumulation in humans with obesity (66). A higher risk of AS is associated with increased oxidant production on account of NO quenching. An imbalance between free radical generation and the antioxidant system can lead to oxidative stress. Increased production of ROS results from the reduced expression of antioxidative enzymes in adipose tissue, including glutathione peroxidase, catalase, and superoxide dismutase.
Oxidative stress linked to atherosclerotic risk factors such as obesity, can also trigger ED (67). According to the oxidative theory of AS, oxidative alteration of lipoproteins causes lipid peroxidation, which, in turn, causes foam cell development and EC destruction in the arterial wall, both of which have a substantial impact on atherogenesis. Furthermore, isoprostanes (IsoPs) contribute to the etiology of vascular dysfunction through their activity. Strong vasoconstrictors in most arterial beds, such as IsoPs, also promote platelet aggregation and increase the adherence of neutrophils and monocytes to ECs, all of which may be involved in AS. Isoprostanes are prostaglandin-like compounds produced by free radical-induced peroxidation independent of cyclo-oxygenase. Furthermore, in individuals with obesity, oxidative stress is involved in various interactions with metabolism and atherogenic lipid activities. These interactions include a postprandial increase in the levels of triglycerides and oxidative modification of LDLs, resulting in the production of oxidized LDL (oxLDL), which has crucial atherogenic properties and a reduction in high-density lipoproteins. These interactions may result in increased ROS production in the endothelium, initiation of proinflammatory vascular processes, and increased endothelial damage, resulting in ED, all of which can lead to the initiation of atherosclerotic processes (68).
In addition to an increase in the levels of oxidative stress markers, such as thiobarbituric acid-reactive substances (TBARS) and protein carbonyls, non–high-density lipoprotein cholesterol (non-HDL-C) levels and the ratios of triglyceride/high-density lipoprotein cholesterol (HDL-C), total cholesterol/HDL-C, and LDL cholesterol (LDL-C)/HDL-C were also increased. In addition, the levels of antioxidant defense markers, such as glutathione (GSH), CAT, and GPx, were reduced (69). Furthermore, increased oxidized LDL levels have been linked to procoagulant and cytotoxic activities, foam cell production, growth, destabilization of atherosclerotic plaques, and increased expression of EC-adhesion molecules. Increased synthesis of vasoconstrictor proteins, oxidative stress, and impaired endothelial function reliant on NO are linked to an increased amount of total adipose tissue and degree of abdominal obesity. Patients who were overweight or affected by obesity had higher levels of phosphorylation of eNOS, catalase, nitrotyrosine, and NADPH oxidase-p47(phox) protein in vascular ECs than patients with normal weight.
A recent review highlighted the association between apolipoprotein M (ApoM), sphingosine-1-phosphate (S1P), HDL, and endothelial dysfunction. The HDL-bound apoM/S1P complex acts as a link between HDL and ECs under normal physiological conditions, preserving the integrity of the endothelial barrier. However, during pathological CV events, this apoM/S1P-signaling leads to inflammation-mediated endothelial dysfunction (70). This could offer a new perspective on the molecular processes that connect obesity to oxidative stress and a higher risk of atherosclerotic CVD (71). Similarly, López-Domènech et al. (72) demonstrated that weight loss results in a decrease in blood pressure, with a significant reduction in the levels of subclinical markers of AS, such as myeloperoxidase (MPO), sP-selectin, small and dense LDL particles, and leukocyte adhesion.
Endothelial dysfunction and vascular calcification
Vascular calcification is involved in vascular remodeling owing to ED, dedifferentiation of VSMCs, and changes in elastin and collagen levels. Medial arterial, intimal, and infantile calcifications are the three primary forms of vascular calcification. Patients with T2DM, chronic renal failure, and advanced age frequently exhibit medial arterial calcification. Nonetheless, intimal calcification is thought to be a passive and degenerative process more commonly associated with AS. It is acknowledged as a self-regulating active process (73). The mechanism of vascular calcification is a tightly regulated, multifactorial process that resembles osteochondrogenesis. It has also been associated with dysregulated metabolism, inflammation, osteogenesis, and progressive AS. The aggressive inflammatory stage of AS is characterized by plaque calcification. Tumor necrosis factor-α and other inflammatory cytokines promote osteogenic differentiation and ECM mineralization. Matrix vesicles are produced by smooth muscle cells and macrophages and may act as sites of initiation for the development of mineral crystals, leading to microcalcifications. Microcalcifications grow into massive masses, spread into the surrounding collagenous matrix from the deeper part of the necrotic core, and ultimately coalesce into calcified sheets or plates (74).
In males, the visceral adiposity index is closely associated with the total coronary calcification score (75). Given that the coronary artery calcium score and total plaque burden are known to be highly correlated, this score can be used to assess the severity of coronary AS.
Impact of human gut microbiota
The gut microbiota (1014 bacteria) plays a vital role in maintaining immune system homeostasis and normal development, regulating the proliferation of epithelial cells, protecting against pathogenic bacteria, and influencing villus architecture and angiogenesis in the intestine (76). Research in both humans and mice have demonstrated that changes in the gut microbiota are linked to several illnesses, such as AS, diabetes mellitus, obesity, and inflammation. The first study in leptin-deficient (ob/ob) mice demonstrated the presence of more phylum Firmicutes and less phylum Bacteroidetes in obese ob/ob mice than in lean controls, and the microbiota have important roles in arterial blood pressure, which is again modulated by the endothelium. Further transfer of the microbiome from obese/lean mice to germ-free mice resulted in the corresponding phenotypes, thereby demonstrating the role of the microbiome in pathology (77, 78). Two main pathways exist by which intestinal bacteria can affect the endothelium of the circulatory system: first, the microbiota and its metabolites can activate the enteric nervous system, which in turn can affect the activity of brain centers that control the cardiovascular system; second, the bacteria can enter the bloodstream through the blood–intestinal barrier and alter the function of the tissues and organs that regulate the circulatory system’s homeostasis. The genus Collinsella is more prevalent in symptomatic atherosclerotic patients, whereas Roseburia and Eubacterium are more prevalent in healthy controls (79). Further metagenomic analysis showed that patient gut metagenomes were deprived of phytoene dehydrogenase and enhanced in genes encoding peptidoglycan synthesis; additionally, patients’ β-carotene serum levels were also decreased. Through fermentation and decomposition, microbiota metabolize resistant starches and dietary fibers, and supply the host with short-chain fatty acids (SCFAs; (acetate, propionate, butyrate). Intestinal epithelial cells, immune cells, and adipocytes have various receptors, including G-protein coupled receptor 41/free fatty acid receptor 3 (GPR41/FFAR3), GPR43/FFAR2, GPR109A, and olfactory receptor 78, to which SFAs bind, despite their varying expression levels between tissues and cell types. Trimethylamine (TMA) and trimethylamine N-oxide (TMAO), metabolites derived from gut microbes, have been shown by Tang et al. to be proatherogenic in both humans and mice (80-82). Aguilar et al. (83, 84) demonstrated using ApoE knockout mice that a butyrate-supplemented chow diet decreased atherosclerotic lesions and further reduced the production of vascular cell adhesion molecule-1, chemotaxis protein-1 (CCL2/MCP-1), and matrix metalloproteinase-2 at the lesion site.
This resulted in increased collagen deposition, reduced macrophage migration, and increased plaque stability. In addition, macrophages from butyrate-treated mice exhibited low ROS and NO release.
The gut microbiota produces TMA, which enters the bloodstream and is oxidized to form TMAO, a cardiac risk biomarker in the liver. Trimethylamine N-oxide has been demonstrated to possess proatherogenic properties owing to ED and the prothrombotic effect caused by platelet aggregation. Activation of the transcription factor NF-kB, which mediates the upregulation of inflammatory signals and adhesion of leukocytes to ECs, results in TMAO-induced ED. Additionally, Seldin et al. demonstrated that CVD and high plasma levels of TMAO are linked to concurrent reduction in endothelial progenitor cells and increased inflammation (85). These studies provide strong evidence that the composition of the gut microbiota and their metabolites play a major role in ED and the resulting AS. Murphy et al. (86) demonstrated that acute infections and other transient stimuli are more likely to cause cumulative arterial damage leading to AS. Disease severity increases with the frequency of acute stimuli and can be made more severe by metabolic, environmental, and genetic risk factors. Animal studies have shown that transient vessel damage heals only partially when the infection is managed. However, repeated inoculation of mice with pathogens significantly enhanced atherosclerotic damage (87) (Fig. 8).
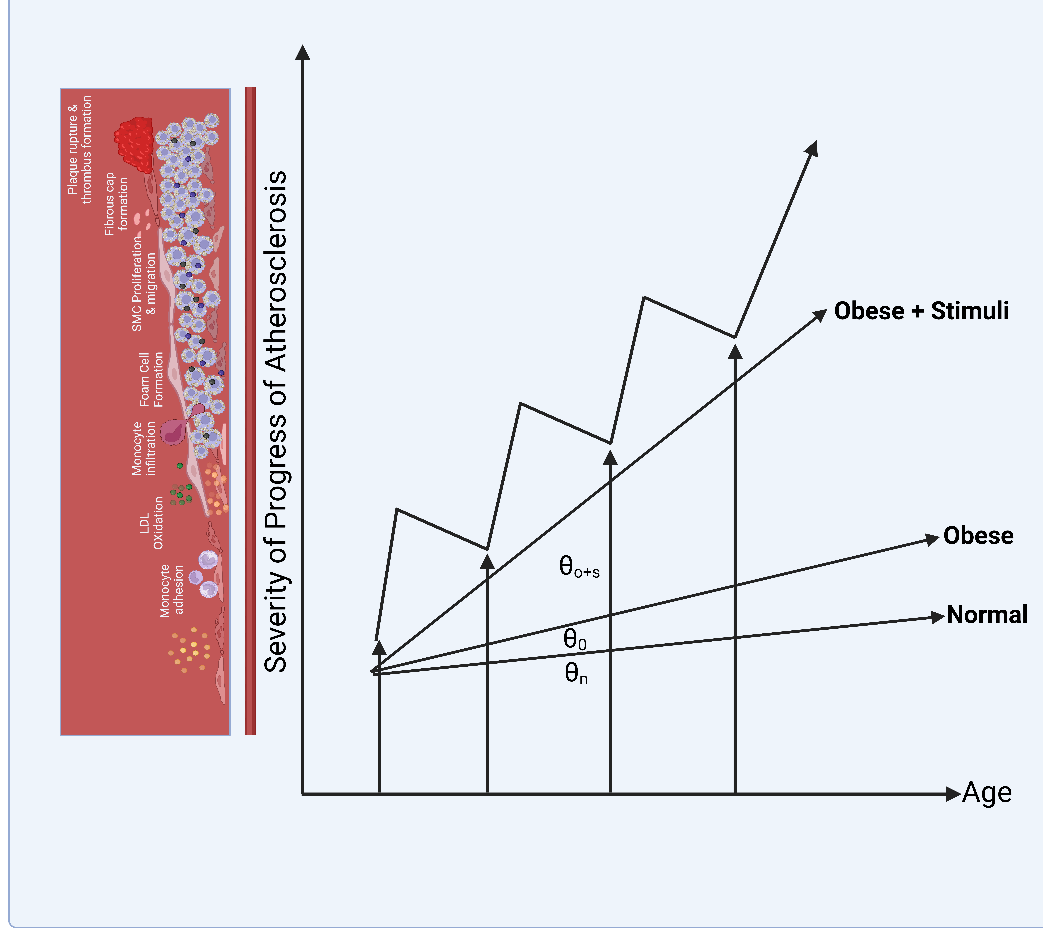
Figure 8. Atherosclerosis development during obesity because of cumulative repetitive infection and inflammatory insult
Reversal of endothelial dysfunction
The reversal of endothelial dysfunction is a promising strategy for preventing ED-mediated diseases. Antioxidants, vitamins, and pharmacological agents have been studied to understand the mechanisms of ED reversal properties (88). It may be possible to reduce the risk of acute CVD events by slowing the progression of AS, altering ED, or both. Free radicals play a major role in age-related endothelial impairment and showed that consumption of an antioxidant cocktail (Vitamin C, 1000 mg; Vitamin E, 600 I.U.; Alpha lipoic acid, 600 mg) restores endothelial function in the elderly, while impairing normal endothelium-dependent vasodilation in the young (89). In addition, various pharmacological agents, such as angiotensin-converting enzyme inhibitors, endothelium NO synthase enhancers, sphingosine-1-phosphate, statins, beta-blockers, phosphodiesterase 5 inhibitors, calcium-channel blockers, and angiotensin AT1 receptor blockers, have endothelial-protective effects (90).
Furthermore, despite their ability to improve glycemic control in humans and animal models of T2DM using dipeptidyl peptidase-4 (DPP4) inhibitors, they also protect against vascular aging through various intricate cellular mechanisms. These include enhancing ED, inhibiting the proliferation and migration of VSMCs, increasing the levels of circulating endothelial progenitor cells (EPCs), and hindering the infiltration of mononuclear macrophages. In addition, DPP4 enhances EC proliferation and migration, mitigates senescence, and blocks apoptosis (91). Recently, the prevention of eNOS uncoupling has attracted considerable interest as a therapeutic option for preventing ED-mediated diseases. One of the most important chemicals secreted by EC is NO, the antiatherogenic properties of which promote cardiovascular homeostasis. Endothelial NO synthase generates vascular NO from the substrate l-arginine (l-Arg), with tetrahydrobiopterin (BH4) serving as the essential cofactor. Under pathological conditions, such as CVD, vascular oxidative stress hinders eNOS activity and favors it’s uncoupling, leading to ED. Uncoupled eNOS generates superoxide anion (O2−) rather than NO, thereby serving as a source of dangerous free radicals that worsen oxidative stress. The key mechanisms underlying eNOS uncoupling include deficiency of the eNOS substrate L-arginine, oxidative depletion of the critical eNOS cofactor BH4, eNOS S-glutathionylation, and accumulation of its analog, asymmetrical dimethylarginine (ADMA). Hence, various pharmacological agents, such as sepiapterin (member of the pteridine class of organic chemicals), 5-methyltetrahydrofolate, fluvastatin, cerivastatin, L-arginine supplementation, and thioredoxin, are being studied to prevent eNOS uncoupling (92). Takase et al. (93) found that ezetimibe in combination with statins improved the functional prognosis and reduced ED in stented coronary arteries. It was also linked to greater reductions in oxysterol levels compared to statin monotherapy (93). Another studies demonstrated that the administration of statins along with eicosatetraenoic acid (EPA), an omega-3 polyunsaturated fatty acid, not only lowers plasma triglyceride levels but also has significant benefits against atherosclerotic plaques (94, 95). The potential benefits of EPA include lowering atherogenic parameters, such as non-HDL-C, oxidized LDL particles, triglycerides, and remnant-like particle cholesterol (RLP-C). It also increases the levels of antiatherogenic factors, thereby improving endothelial function, enhancing antioxidant properties, and secreting pro-resolving lipid mediators called resolvins (94, 96). Furthermore, they decrease macrophage accumulation, monocyte adhesion, and foam cell accumulation. These studies provide strong evidence that ED reversal holds promising therapeutic potential to prevent or at least delay AS.
Conclusion and future directions
Although it is difficult to isolate the specific mechanism underlying obesity-mediated AS, alterations in endothelial structure and function, cellular senescence, epigenetic factors, oxidative stress, vascular calcification, and gut microbiome play crucial roles in disease incidence and progression. Exploring the potential link between these factors not only helps to understand the mechanisms of the disease but also to find a multi-targeted therapeutic agent to prevent or slow down the AS process. Targeting these mechanisms through both prevention and treatment at an early stage can delay the progression of AS. Further lifestyle and diet changes are keys to prevent obesity in the first place.
Peer-review: External and internal
Conflict of interest: None to declare
Authorship: Both authors have made substantial contributions to the scientific quality of this paper by fulfilling the ICMJE criteria. Conceptualization and project administration: E. H. A. Writing the first draft: R. L. J. Writing–review & editing: R. L. J. & E.H.A. The authors are accountable for all the aspects of this review.
Acknowledgement and funding: None to declare
Statement on A.I.-assisted technologies use: We declare that we did not use AI-assisted technologies in preparation of this manuscript
Availability of data and material: Not applies
References
| 1.Nagpal N, Messito MJ, Katzow M, Gross RS. Obesity in children. Pediatrics Rev 2022; 43: 601-17. doi: 10.1542/pir.2021-005095 https://doi.org/10.1542/pir.2021-005095 PMid:36316265 |
||||
| 2.Kozieł P, Jankowski P, Mirek-Bryniarska E, Nessler J, Podolec P, De Bacquer D, et al. Obesity in patients with established coronary artery disease over a 20-year period (1997-2017). Polish Arch Int Med 2021; 131: 26-32. doi: 10.20452/pamw.15742 https://doi.org/10.20452/pamw.15742 |
||||
| 3. Azab M, Al-Shudifat AE, Johannessen A, Al-Shdaifat A, Agraib LM, Tayyem RF. Are risk factors for coronary artery disease different in persons with and without obesity? Metab Syndr Rel Disord 2018; 16: 440-5. doi: 10.1089/met.2017.0152 https://doi.org/10.1089/met.2017.0152 PMid:30088947 |
||||
| 4.Khan SS, Ning H, Wilkins JT, Allen N, Carnethon M, Berry JD, et al. Association of body mass index with lifetime risk of cardiovascular disease and compression of morbidity. JAMA Cardiol 2018; 3: 280-7. doi: 10.1001/jamacardio.2018.0022 https://doi.org/10.1001/jamacardio.2018.0022 PMid:29490333 PMCid:PMC5875319 |
||||
| 5.Kajikawa M, Maruhashi T, Kishimoto S, Hashimoto H, Takaeko Y, Yamaji T, et al. Association of body mass index with endothelial function in Asian men. Int J Cardiol 2021; 324: 186-92. doi: 10.1016/j.ijcard.2020.09.029 https://doi.org/10.1016/j.ijcard.2020.09.029 PMid:32931855 |
||||
| 6.Dai H, Alsalhe TA, Chalghaf N, Riccò M, Bragazzi NL, Wu J. The global burden of disease attributable to high body mass index in 195 countries and territories, 1990-2017: An analysis of the Global Burden of Disease Study. PLoS Med 2020; 17: e1003198. doi: 10.1371/journal.pmed.1003198. https://doi.org/10.1371/journal.pmed.1003198 PMid:32722671 PMCid:PMC7386577 |
||||
| 7.Lassale C, Tzoulaki I, Moons KGM, Sweeting M, Boer J, Johnson L, et al. Separate and combined associations of obesity and metabolic health with coronary heart disease: a pan-European case-cohort analysis. Eur Heart J 2018; 39: 397-406. doi: 10.1093/eurheartj/ehx448. https://doi.org/10.1093/eurheartj/ehx448 PMid:29020414 PMCid:PMC6198928 |
||||
| 8.Yoon JW, Jung CH, Kim MK, Park HE, Park KS, Jang HC, et al. Influence of the definition of "metabolically healthy obesity" on the progression of coronary artery calcification. PloS One 2017; 12: e0178741. doi: 10.1371/journal.pone.0178741 https://doi.org/10.1371/journal.pone.0178741 PMid:28575097 PMCid:PMC5456095 |
||||
| 9.Kajikawa M, Higashi Y. Obesity and endothelial function. Biomedicines 2022; 10. doi: 10.3390/biomedicines10071745 https://doi.org/10.3390/biomedicines10071745 PMid:35885049 PMCid:PMC9313026 |
||||
| 10.Sena CM, Pereira AM, Seiça R. Endothelial dysfunction - a major mediator of diabetic vascular disease. Biochim Biophys Acta 2013; 1832: 2216-31. doi: 10.1016/j.bbadis.2013.08.006 https://doi.org/10.1016/j.bbadis.2013.08.006 PMid:23994612 |
||||
| 11.McGill HC, Jr. Fatty streaks in the coronary arteries and aorta. Lab Investigat: J Tech Meth Pathol 1968; 18: 560-4. | ||||
| 12.Strong JP, Malcom GT, McMahan CA, Tracy RE, Newman WP, 3rd, Herderick EE, et al. Prevalence and extent of atherosclerosis in adolescents and young adults: implications for prevention from the Pathobiological Determinants of Atherosclerosis in Youth Study. JAMA 1999; 281: 727-35.. doi: 10.1001/jama.281.8.727 https://doi.org/10.1001/jama.281.8.727 PMid:10052443 |
||||
| 13.Virdis A, Santini F, Colucci R, Duranti E, Salvetti G, Rugani I, et al. Vascular generation of tumor necrosis factor-α reduces nitric oxide availability in small arteries from visceral fat of obese patients. J Am Coll Cardiol 2011; 58: 238-47. doi: 10.1016/j.jacc.2011.01.050 https://doi.org/10.1016/j.jacc.2011.01.050 PMid:21737013 |
||||
| 14.Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab 2004; 89: 2548-56. doi: 10.1210/jc.2004-0395 https://doi.org/10.1210/jc.2004-0395 PMid:15181022 |
||||
| 15.Fancher IS, Le Master E, Ahn SJ, Adamos C, Lee JC, Berdyshev E, et al. Impairment of flow-sensitive inwardly rectifying K(+) channels via disruption of glycocalyx mediates obesity-induced endothelial dysfunction. Arterioscl Thromb Vasc Biol 2020; 40: e240-e55. doi: 10.1161/atvbaha.120.314935 https://doi.org/10.1161/ATVBAHA.120.314935 PMid:32698687 PMCid:PMC7503211 |
||||
| 16.Xu J, Zou MH. Molecular insights and therapeutic targets for diabetic endothelial dysfunction. Circulation 2009; 120: 1266-86. doi: 10.1161/circulationaha.108.835223 https://doi.org/10.1161/CIRCULATIONAHA.108.835223 PMid:19786641 PMCid:PMC2910587 |
||||
| 17.Barton M. Obesity and aging: determinants of endothelial cell dysfunction and atherosclerosis. Pflugers Arch: Eur J Physiol 2010; 460: 825-37. doi: 10.1007/s00424-010-0860-y https://doi.org/10.1007/s00424-010-0860-y PMid:20635093 |
||||
| 18.Kwaifa IK, Bahari H, Yong YK, Noor SM. Endothelial dysfunction in obesity-induced inflammation: molecular mechanisms and clinical implications. Biomolecules 2020; 10. doi: 10.3390/biom10020291 https://doi.org/10.3390/biom10020291 PMid:32069832 PMCid:PMC7072669 |
||||
| 19.Chen J, Jiang L, Yu XH, Hu M, Zhang YK, Liu X, et al. Endocan: A key player of cardiovascular disease. Front Cardiovasc Med 2021; 8: 798699. doi: 10.3389/fcvm.2021.798699 https://doi.org/10.3389/fcvm.2021.798699 PMid:35071362 PMCid:PMC8766991 |
||||
| 20.Li CG, Bethell H, Wilson PB, Bhatnagar D, Walker MG, Kumar S. The significance of CD105, TGFbeta and CD105/TGFbeta complexes in coronary artery disease. Atherosclerosis 2000; 152: 249-56. doi: 10.1016/s0021-9150(99)00476-1 https://doi.org/10.1016/S0021-9150(99)00476-1 PMid:10996361 |
||||
| 21.Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nature Rev Immunol 2006; 6: 772-83. doi: 10.1038/nri1937 https://doi.org/10.1038/nri1937 PMid:16998510 |
||||
| 22.Cypess AM. Reassessing human adipose tissue. New Engl J Med 2022; 386: 768-79. doi: 10.1056/NEJMra2032804 https://doi.org/10.1056/NEJMra2032804 PMid:35196429 |
||||
| 23.Darvall KA, Sam RC, Silverman SH, Bradbury AW, Adam DJ. Obesity and Thrombosis. Eur J Vasc Endovasc Surg 2007; 33: 223-33. doi: 10.1016/j.ejvs.2006.10.006 https://doi.org/10.1016/j.ejvs.2006.10.006 PMid:17185009 |
||||
| 24.O'Mara AE, Johnson JW, Linderman JD, Brychta RJ, McGehee S, Fletcher LA, et al. Chronic mirabegron treatment increases human brown fat, HDL cholesterol, and insulin sensitivity. J Clin Invest 2020; 130: 2209-19. doi: 10.1172/jci131126 https://doi.org/10.1172/JCI131126 PMid:31961826 PMCid:PMC7190915 |
||||
| 25.Karastergiou K, Mohamed-Ali V. The autocrine and paracrine roles of adipokines. Mol Cell Endocrin 2010; 318: 69-78. doi: 10.1016/j.mce.2009.11.011 https://doi.org/10.1016/j.mce.2009.11.011 PMid:19948207 |
||||
| 26.Yang X, Yi X, Zhang F, Li F, Lang L, Ling M, et al. Cytochrome P450 epoxygenase-derived EPA and DHA oxylipins 17,18-epoxyeicosatetraenoic acid and 19,20-epoxydocosapentaenoic acid promote BAT thermogenesis and WAT browning through the GPR120-AMPKα signaling pathway. Food Funct 2022; 13: 1232-45. doi: 10.1039/d1fo02608a https://doi.org/10.1039/D1FO02608A PMid:35019933 |
||||
| 27.Segovia SA, Vickers MH, Gray C, Reynolds CM. Maternal obesity, inflammation, and developmental programming. BioMed Res Int 2014; 2014: 418975. doi: 10.1155/2014/418975 https://doi.org/10.1155/2014/418975 PMid:24967364 PMCid:PMC4055365 |
||||
| 28.Thompson D, Karpe F, Lafontan M, Frayn K. Physical activity and exercise in the regulation of human adipose tissue physiology. Physiol Rev 2012; 92: 157-91. doi: 10.1152/physrev.00012.2011 https://doi.org/10.1152/physrev.00012.2011 PMid:22298655 |
||||
| 29.Rosenquist KJ, Pedley A, Massaro JM, Therkelsen KE, Murabito JM, Hoffmann U, et al. Visceral and subcutaneous fat quality and cardiometabolic risk. JACC Cardiovasc Imag 2013; 6: 762-71. doi: 10.1016/j.jcmg.2012.11.021 https://doi.org/10.1016/j.jcmg.2012.11.021 PMid:23664720 PMCid:PMC3745280 |
||||
| 30.Park HS, Park JY, Yu R. Relationship of obesity and visceral adiposity with serum concentrations of CRP, TNF-alpha and IL-6. Diabetes Res Clin Pract 2005; 69: 29-35. doi: 10.1016/j.diabres.2004.11.007 https://doi.org/10.1016/j.diabres.2004.11.007 PMid:15955385 |
||||
| 31.Gealekman O, Guseva N, Hartigan C, Apotheker S, Gorgoglione M, Gurav K, et al. Depot-specific differences and insufficient subcutaneous adipose tissue angiogenesis in human obesity. Circulation 2011; 123: 186-94. doi: 10.1161/circulationaha.110.970145 https://doi.org/10.1161/CIRCULATIONAHA.110.970145 PMid:21200001 PMCid:PMC3334340 |
||||
| 32.Alvehus M, Burén J, Sjöström M, Goedecke J, Olsson T. The human visceral fat depot has a unique inflammatory profile. Obesity 2010; 18: 879-83. doi: 10.1038/oby.2010.22 https://doi.org/10.1038/oby.2010.22 PMid:20186138 |
||||
| 33.Ledoux S, Queguiner I, Msika S, Calderari S, Rufat P, Gasc JM, et al. Angiogenesis associated with visceral and subcutaneous adipose tissue in severe human obesity. Diabetes 2008; 57: 3247-57. doi: 10.2337/db07-1812 https://doi.org/10.2337/db07-1812 PMid:18835936 PMCid:PMC2584130 |
||||
| 34.Jaffe EA. Cell biology of endothelial cells. Human Pathol 1987; 18: 234-9. doi: 10.1016/s0046-8177(87)80005-9 https://doi.org/10.1016/S0046-8177(87)80005-9 PMid:3546072 |
||||
| 35.Luft JH. Fine structures of capillary and endocapillary layer as revealed by ruthenium red. Federation proceedings. 1966;25(6):1773-83. Epub 1966/11/01. PubMed PMID: 5927412. | ||||
| 36.Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288(5789):373-6. Epub 1980/11/27. doi: 10.1038/288373a0. PubMed PMID: 6253831. https://doi.org/10.1038/288373a0 PMid:6253831 |
||||
| 37.Goligorsky MS. Endothelial cell dysfunction: can't live with it, how to live without it. Am J Physiol Renal Physiol 2005; 288: F871-80. doi: 10.1152/ajprenal.00333.20047 https://doi.org/10.1152/ajprenal.00333.2004 PMid:15821252 |
||||
| 38.Félétou M, Vanhoutte PM. Endothelial dysfunction: a multifaceted disorder (The Wiggers Award Lecture). Am J Physiol Heart Circ Physiol 2006; 291: H985-1002. doi: 10.1152/ajpheart.00292.2006 https://doi.org/10.1152/ajpheart.00292.2006 PMid:16632549 |
||||
| 39.Reitsma S, Slaaf DW, Vink H, van Zandvoort MA, oude Egbrink MG. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch: Eur J Physiol 2007; 454: 345-59. doi: 10.1007/s00424-007-0212-8 https://doi.org/10.1007/s00424-007-0212-8 PMid:17256154 PMCid:PMC1915585 |
||||
| 40.Mussbacher M, Salzmann M, Brostjan C, Hoesel B, Schoergenhofer C, Datler H, et al. Cell type-specific roles of NF-ΚB linking inflammation and thrombosis. Front Immunol 2019; 10: 85. doi: 10.3389/fimmu.2019.00085 https://doi.org/10.3389/fimmu.2019.00085 PMid:30778349 PMCid:PMC6369217 |
||||
| 41.Katakami N. Mechanism of development of atherosclerosis and cardiovascular disease in diabetes mellitus. J Atheroscler Thromb 2018; 25: 27-39. doi: 10.5551/jat.RV17014 https://doi.org/10.5551/jat.RV17014 PMid:28966336 PMCid:PMC5770221 |
||||
| 42.Donato AJ, Machin DR, Lesniewski LA. Mechanisms of dysfunction in the aging vasculature and role in age-related disease. Circ Res 2018; 123: 825-48. doi: 10.1161/circresaha.118.312563 https://doi.org/10.1161/CIRCRESAHA.118.312563 PMid:30355078 PMCid:PMC6207260 |
||||
| 43.Zhan H, Suzuki T, Aizawa K, Miyagawa K, Nagai R. Ataxia telangiectasia mutated (ATM)-mediated DNA damage response in oxidative stress-induced vascular endothelial cell senescence. J Biol Chem 2010; 285: 29662-70. doi: 10.1074/jbc.M110.125138 https://doi.org/10.1074/jbc.M110.125138 PMid:20639198 PMCid:PMC2937997 |
||||
| 44.Spyridopoulos I, Isner JM, Losordo DW. Oncogenic ras induces premature senescence in endothelial cells: role of p21(Cip1/Waf1). Basic Res Cardiol 2002; 97: 117-24. doi: 10.1007/s003950200001 https://doi.org/10.1007/s003950200001 PMid:12002258 |
||||
| 45.Dai DF, Rabinovitch PS, Ungvari Z. Mitochondria and cardiovascular aging. Circ Res 2012; 110: 1109-24. doi: 10.1161/circresaha.111.246140 https://doi.org/10.1161/CIRCRESAHA.111.246140 PMid:22499901 PMCid:PMC3867977 |
||||
| 46.Jia G, Aroor AR, DeMarco VG, Martinez-Lemus LA, Meininger GA, Sowers JR. Vascular stiffness in insulin resistance and obesity. Front Physiol 2015; 6: 231. doi: 10.3389/fphys.2015.00231 https://doi.org/10.3389/fphys.2015.00231 |
||||
| 47.Donato AJ, Morgan RG, Walker AE, Lesniewski LA. Cellular and molecular biology of aging endothelial cells. J Mol Cell Cardiol 2015; 89: 122-35. doi: 10.1016/j.yjmcc.2015.01.021 https://doi.org/10.1016/j.yjmcc.2015.01.021 PMid:25655936 PMCid:PMC4522407 |
||||
| 48.Vasan RS, Beiser A, Seshadri S, Larson MG, Kannel WB, D'Agostino RB, et al. Residual lifetime risk for developing hypertension in middle-aged women and men: The Framingham Heart Study. JAMA 2002; 287: 1003-10. doi: 10.1001/jama.287.8.1003 https://doi.org/10.1001/jama.287.8.1003 PMid:11866648 |
||||
| 49.Zhou J, Wang Q, Ding Y, Zou MH. Hypochlorous acid via peroxynitrite activates protein kinase Cθ and insulin resistance in adipocytes. J Mol Endocrinol 2015; 54: 25-37. doi: 10.1530/jme-14-0213. https://doi.org/10.1530/JME-14-0213 PMid:25381390 PMCid:PMC4261204 |
||||
| 50.Jia G, Habibi J, Aroor AR, Hill MA, DeMarco VG, Lee LE, et al. Enhanced endothelium epithelial sodium channel signaling prompts left ventricular diastolic dysfunction in obese female mice. Metabolism: Clin Exper 2018; 78: 69-79. doi: 10.1016/j.metabol.2017.08.008 https://doi.org/10.1016/j.metabol.2017.08.008 PMid:28920862 |
||||
| 51.Briley-Saebo KC, Shaw PX, Mulder WJ, Choi SH, Vucic E, Aguinaldo JG, et al. Targeted molecular probes for imaging atherosclerotic lesions with magnetic resonance using antibodies that recognize oxidation-specific epitopes. Circulation 2008; 117: 3206-15. doi: 10.1161/circulationaha.107.757120 https://doi.org/10.1161/CIRCULATIONAHA.107.757120 PMid:18541740 PMCid:PMC4492476 |
||||
| 52.Bochkov VN. Inflammatory profile of oxidized phospholipids. Thromb Hemost 2007; 97: 348-54. https://doi.org/10.1160/TH06-08-0474 |
||||
| 53.Pal PB, Sonowal H, Shukla K, Srivastava SK, Ramana KV. Aldose reductase mediates NLRP3 inflammasome-initiated innate immune response in hyperglycemia-induced Thp1 monocytes and male mice. Endocrinol 2017; 158: 3661-75. doi: 10.1210/en.2017-00294 https://doi.org/10.1210/en.2017-00294 PMid:28938395 PMCid:PMC5659696 |
||||
| 54.Khan SY, Awad EM, Oszwald A, Mayr M, Yin X, Waltenberger B, et al. Premature senescence of endothelial cells upon chronic exposure to TNFα can be prevented by N-acetyl cysteine and plumericin. Sci Rep 2017; 7: 39501. doi: 10.1038/srep39501 https://doi.org/10.1038/srep39501 PMid:28045034 PMCid:PMC5206708 |
||||
| 55.Poznyak AV, Nikiforov NG, Markin AM, Kashirskikh DA, Myasoedova VA, Gerasimova EV, et al. Overview of oxLDL and its impact on cardiovascular health: Focus on atherosclerosis. Front Pharmacol 2020; 11 :613780. doi: 10.3389/fphar.2020.613780 https://doi.org/10.3389/fphar.2020.613780 PMid:33510639 PMCid:PMC7836017 |
||||
| 56.Kovanen PT. Mast cells as potential accelerators of human atherosclerosis-from early to late lesions. Int J Mol Sci 2019; 20. doi: 10.3390/ijms20184479 https://doi.org/10.3390/ijms20184479 PMid:31514285 PMCid:PMC6770933 |
||||
| 57.Canepa M, Viazzi F, Strait JB, Ameri P, Pontremoli R, Brunelli C, et al. Longitudinal association between serum uric acid and arterial stiffness: results from the baltimore longitudinal study of aging. Hypertension 2017; 69: 228-35. doi: 10.1161/hypertensionaha.116.08114 https://doi.org/10.1161/HYPERTENSIONAHA.116.08114 PMid:27956574 PMCid:PMC5354105 |
||||
| 58.Aroor AR, Jia G, Habibi J, Sun Z, Ramirez-Perez FI, Brady B, et al. Uric acid promotes vascular stiffness, maladaptive inflammatory responses and proteinuria in western diet fed mice. Metabolism: Clin Exp 2017; 74: 32-40. doi: 10.1016/j.metabol.2017.06.006 https://doi.org/10.1016/j.metabol.2017.06.006 PMid:28764846 PMCid:PMC5577816 |
||||
| 59.Neogi T, Ellison RC, Hunt S, Terkeltaub R, Felson DT, Zhang Y. Serum uric acid is associated with carotid plaques: the National Heart, Lung, and Blood Institute Family Heart Study. J Rheumatol 2009; 36: 378-84. doi: 10.3899/jrheum.080646 https://doi.org/10.3899/jrheum.080646 PMid:19012359 PMCid:PMC2731484 |
||||
| 60.Han MS, Jung DY, Morel C, Lakhani SA, Kim JK, Flavell RA, et al. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science 2013; 339: 218-22. doi: 10.1126/science.1227568 https://doi.org/10.1126/science.1227568 PMid:23223452 PMCid:PMC3835653 |
||||
| 61.Bolton E, Rajkumar C. The ageing cardiovascular system. Rev Clin Gerontol 2011; 21: 99-109. doi: 10.1017/S0959259810000389 https://doi.org/10.1017/S0959259810000389 |
||||
| 62.Lund G, Zaina S. Atherosclerosis: an epigenetic balancing act that goes wrong. Curr Atheroscl Rep 2011; 13: 208-14. doi: 10.1007/s11883-011-0174-3 https://doi.org/10.1007/s11883-011-0174-3 PMid:21384259 |
||||
| 63.Beekman M, Blanché H, Perola M, Hervonen A, Bezrukov V, Sikora E, et al. Genome‐wide linkage analysis for human longevity: Genetics of Healthy Aging Study. Aging cell 2013; 12: 184-93. https://doi.org/10.1111/acel.12039 PMid:23286790 PMCid:PMC3725963 |
||||
| 64.Kurz DJ, Kloeckener-Gruissem B, Akhmedov A, Eberli FR, Bühler I, Berger W, et al. Degenerative aortic valve stenosis, but not coronary disease, is associated with shorter telomere length in the elderly. Arterioscler Thromb Vasc Biol 2006; 26: e114-e7. https://doi.org/10.1161/01.ATV.0000222961.24912.69 PMid:16627805 |
||||
| 65.Bianchessi V, Badi I, Bertolotti M, Nigro P, D'Alessandra Y, Capogrossi MC, et al. The mitochondrial lncRNA ASncmtRNA-2 is induced in aging and replicative senescence in endothelial cells. J Mol Cell Cardiol 2015; 81: 62-70. https://doi.org/10.1016/j.yjmcc.2015.01.012 PMid:25640160 |
||||
| 66.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 2017; 114: 1752-61. https://doi.org/10.1172/JCI21625 PMid:15599400 PMCid:PMC535065 |
||||
| 67.Chisolm GM, Steinberg D. The oxidative modification hypothesis of atherogenesis: an overview. Free Rad Biol Med 2000; 28: 1815-26. https://doi.org/10.1016/S0891-5849(00)00344-0 PMid:10946223 |
||||
| 68.Matsuda M, Shimomura I. Increased oxidative stress in obesity: implications for metabolic syndrome, diabetes, hypertension, dyslipidemia, atherosclerosis, and cancer. Obes Res Clin Pract 2013; 7: e330-e41. https://doi.org/10.1016/j.orcp.2013.05.004 PMid:24455761 |
||||
| 69.Nanda N, Bobby Z, Hamide A, Koner BC, Sridhar MG. Association between oxidative stress and coronary lipid risk factors in hypothyroid women is independent of body mass index. Metabol Clin Exp 2007; 56: 1350-5. https://doi.org/10.1016/j.metabol.2007.05.015 PMid:17884444 |
||||
| 70.Mattisson IY, Christoffersen C. Apolipoprotein M and its impact on endothelial dysfunction and inflammation in the cardiovascular system. Atheroscler 2021; 334: 76-84. https://doi.org/10.1016/j.atherosclerosis.2021.08.039 PMid:34482091 |
||||
| 71.Silver AE, Beske SD, Christou DD, Donato AJ, Moreau KL, Eskurza I, et al. Overweight and obese humans demonstrate increased vascular endothelial NAD (P) H oxidase-p47phox expression and evidence of endothelial oxidative stress. Circulation 2007; 115: 627-37. https://doi.org/10.1161/CIRCULATIONAHA.106.657486 PMid:17242275 |
||||
| 72.López-Domènech S, Martínez-Herrera M, Abad-Jiménez Z, Morillas C, Escribano-López I, Díaz-Morales N, et al. Dietary weight loss intervention improves subclinical atherosclerosis and oxidative stress markers in leukocytes of obese humans. Int J Obes 2019; 43: 2200-9. https://doi.org/10.1038/s41366-018-0309-5 PMid:30622308 |
||||
| 73.Amizuka N, Hasegawa T, Oda K, de Freitas PHL, Hoshi K, Li M, et al. Histology of epiphyseal cartilage calcification and endochondral ossification. Front Bioscience-Elite 2012; 4: 2085-100. https://doi.org/10.2741/e526 PMid:22202021 |
||||
| 74.Shi X, Gao J, Lv Q, Cai H, Wang F, Ye R, et al. Calcification in atherosclerotic plaque vulnerability: Friend or foe? Front Physiol 2020; 11: 56. doi: 10.3389/fphys.2020.00056 https://doi.org/10.3389/fphys.2020.00056 PMid:32116766 PMCid:PMC7013039 |
||||
| 75.Bagyura Z, Kiss L, Lux Á, Csobay-Novák C, Jermendy Á L, Polgár L, et al. Association between coronary atherosclerosis and visceral adiposity index. Nutr Metabol Cardiovasc Dis 2020; 30: 796-803. doi: 10.1016/j.numecd.2020.01.013 https://doi.org/10.1016/j.numecd.2020.01.013 PMid:32127334 |
||||
| 76.Sommer F, Bäckhed F. The gut microbiota-masters of host development and physiology. Nat Rev Microbiol 2013; 11: 227-38. https://doi.org/10.1038/nrmicro2974 PMid:23435359 |
||||
| 77.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature 2009; 457: 480-4. doi: 10.1038/nature07540 https://doi.org/10.1038/nature07540 PMid:19043404 PMCid:PMC2677729 |
||||
| 78.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Nat Acad Sci USA 2008; 105: 16731-6. doi: 10.1073/pnas.0804812105 https://doi.org/10.1073/pnas.0804812105 PMid:18936492 PMCid:PMC2575488 |
||||
| 79.Karlsson FH, Tremaroli V, Nookaew I, Bergström G, Behre CJ, Fagerberg B, et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013; 498: 99-103. doi: 10.1038/nature12198 80.Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, DuGar B, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011; 472: 57-63. https://doi.org/10.1038/nature09922 PMid:21475195 PMCid:PMC3086762 |
||||
| 81.Tang WW, Hazen SL. The contributory role of gut microbiota in cardiovascular disease. J Clin Invest 2014; 124: 4204-11. https://doi.org/10.1172/JCI72331 PMid:25271725 PMCid:PMC4215189 |
||||
| 82.Tang WW, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. New Engl J Med 2013; 368: 1575-84. https://doi.org/10.1056/NEJMoa1109400 PMid:23614584 PMCid:PMC3701945 |
||||
| 83.Aguilar EC, Leonel AJ, Teixeira LG, Silva AR, Silva JF, Pelaez JM, et al. Butyrate impairs atherogenesis by reducing plaque inflammation and vulnerability and decreasing NFκB activation. Nutr Metabol Cardiovasc Dis 2014; 24: 606-13. doi: 10.1016/j.numecd.2014.01.002 https://doi.org/10.1016/j.numecd.2014.01.002 PMid:24602606 |
||||
| 84.Aguilar EC, Santos LC, Leonel AJ, de Oliveira JS, Santos EA, Navia-Pelaez JM, et al. Oral butyrate reduces oxidative stress in atherosclerotic lesion sites by a mechanism involving NADPH oxidase down-regulation in endothelial cells. J Nutr Biochem 2016; 34: 99-105. doi: 10.1016/j.jnutbio.2016.05.002 https://doi.org/10.1016/j.jnutbio.2016.05.002 PMid:27261536 |
||||
| 85.Seldin MM, Meng Y, Qi H, Zhu W, Wang Z, Hazen SL, et al. Trimethylamine N-oxide promotes vascular inflammation through signaling of mitogen-activated protein kinase and nuclear factor-κB. J Am Heart Assoc 2016; 5. doi: 10.1161/jaha.115.002767 https://doi.org/10.1161/JAHA.115.002767 PMid:26903003 PMCid:PMC4802459 |
||||
| 86.Murphy EA. Some difficulties in the investigation of genetic factors in coronary artery disease. Can Med Assoc J 1967; 97: 1181-92. | ||||
| 87.Libby P, Egan D, Skarlatos S. Roles of infectious agents in atherosclerosis and restenosis: an assessment of the evidence and need for future research. Circulation 1997; 96: 4095-103. doi: 10.1161/01.cir.96.11.4095 https://doi.org/10.1161/01.CIR.96.11.4095 PMid:9403635 |
||||
| 88.Ulker S, McKeown PP, Bayraktutan U. Vitamins reverse endothelial dysfunction through regulation of eNOS and NAD(P)H oxidase activities. Hypertension 2003; 41: 534-9. doi: 10.1161/01.hyp.0000057421.28533.37 https://doi.org/10.1161/01.HYP.0000057421.28533.37 PMid:12623955 |
||||
| 89.Wray DW, Nishiyama SK, Harris RA, Zhao J, McDaniel J, Fjeldstad AS, et al. Acute reversal of endothelial dysfunction in the elderly after antioxidant consumption. Hypertension 2012; 59: 818-24. doi: 10.1161/hypertensionaha.111.189456 https://doi.org/10.1161/HYPERTENSIONAHA.111.189456 PMid:22353612 PMCid:PMC3321727 |
||||
| 90.Su JB. Vascular endothelial dysfunction and pharmacological treatment. World J Cardiol 2015; 7: 719-41. doi: 10.4330/wjc.v7.i11.719 https://doi.org/10.4330/wjc.v7.i11.719 PMid:26635921 PMCid:PMC4660468 |
||||
| 91.Cao F, Wu K, Zhu YZ, Bao ZW. Roles and mechanisms of dipeptidyl peptidase 4 inhibitors in vascular aging. Front Endocrinol 2021; 12: 731273. doi: 10.3389/fendo.2021.731273 https://doi.org/10.3389/fendo.2021.731273 PMid:34489872 PMCid:PMC8416540 |
||||
| 92.Janaszak-Jasiecka A, Płoska A, Wierońska JM, Dobrucki LW, Kalinowski L. Endothelial dysfunction due to eNOS uncoupling: molecular mechanisms as potential therapeutic targets. Cell Mol Biol Lett 2023; 28: 21. doi: 10.1186/s11658-023-00423-2 https://doi.org/10.1186/s11658-023-00423-2 PMid:36890458 PMCid:PMC9996905 |
||||
| 93.Takase S, Matoba T, Nakashiro S, Mukai Y, Inoue S, Oi K, et al. Ezetimibe in combination with statins ameliorates endothelial dysfunction in coronary arteries after stenting: The CuVIC Trial (Effect of Cholesterol Absorption Inhibitor Usage on Target Vessel Dysfunction After Coronary Stenting), a multicenter randomized controlled trial. Arterioscler Thromb Vasc Biol 2017;37(2):350-8. doi: 10.1161/atvbaha.116.308388 https://doi.org/10.1161/ATVBAHA.116.308388 PMid:27932353 |
||||
| 94.Patel AA, Budoff MJ. Effects of eicosapentaenoic acid and docosahexaenoic acid on lipoproteins in hypertriglyceridemia. Curr Opin Endocrinol Diabet Obes 2016; 23: 145-9. doi: 10.1097/med.0000000000000233 https://doi.org/10.1097/MED.0000000000000233 PMid:26825470 |
||||
| 95.Nelson JR, Wani O, May HT, Budoff M. Potential benefits of eicosapentaenoic acid on atherosclerotic plaques. Vasc Pharmacol 2017; 91: 1-9. doi: 10.1016/j.vph.2017.02.004 https://doi.org/10.1016/j.vph.2017.02.004 PMid:28263852 |
||||
| 96.Borow KM, Nelson JR, Mason RP. Biologic plausibility, cellular effects, and molecular mechanisms of eicosapentaenoic acid (EPA) in atherosclerosis. Atheroscler 2015; 242: 357-66. doi: 10.1016/j.atherosclerosis.2015.07.035 https://doi.org/10.1016/j.atherosclerosis.2015.07.035 PMid:26253795 |
||||
Copyright

This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.
AUTHOR'S CORNER
